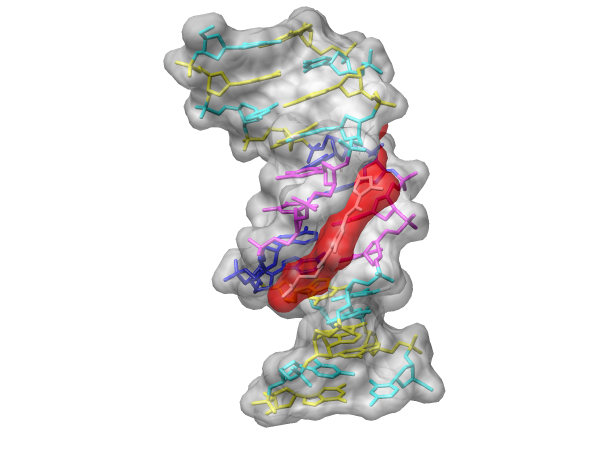
DNA Helix with bound Netropsin (6bna.pdb)
DNA Helix with bound Netropsin (6bna.pdb)
The purpose of this tutorial is to provide a basic overview of Chimera and some of the basic commands and features for displaying and manipulating Protein Data Bank (PDB) models. Chimera provides two basic modes of interaction: menus and command-line. All of the basic functionality in Chimera is available through both forms of interaction, but extensions are often only available through the menu interface, and some commands and the scripting capability are not available through any of the menus. For this reason it is suggested that users become familiar with both ways of interacting with Chimera.
To begin working with the tutorial, you will need to have access to the PDB files 1zik.pdb and 6bna.pdb, which are provided with the Chimera distribution. If you have Internet access, these files may be fetched directly from the Protein Data Bank.
| Item | Example | Description |
| Keyboard key | Ctrl | The control key |
| Mouse key | Btn1 | Mouse button 1 (left button) |
| Menu action | File→Open | File Menu bar pulldown, followed by Open |
| Filename (or file path) | 1zik.pdb | File 1zik.pdb |
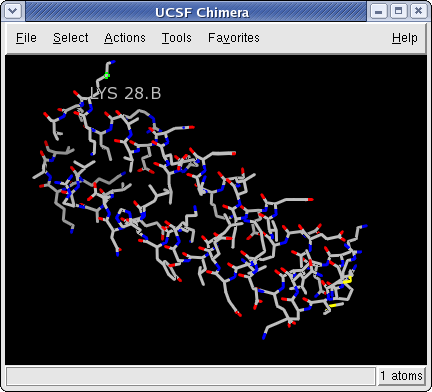
UCSF Chimera with 1zik.pdb loaded
On Linux run the executable "chimera" in the bin directory of your Chimera installation. If Chimera is installed in /usr/local/chimera, then you run /usr/local/chimera/bin/chimera from a shell.
On Windows start Chimera by double clicking the Chimera icon in the directory called bin in your Chimera installation. If Chimera is installed in \Program Files then the executable will be found in directory \Program Files\Chimera\bin. A Chimera icon will also be put on your desktop by default.
On Mac start the Apple X server found in /Applications/Utilities/X11. Then double click the Chimera application to start. The X server is not installed in Mac OS 10.3 by default and if you do not have it you can download it from Apple. It is necessary for running Chimera on the Mac.
A basic Chimera window should appear after a few seconds. Chimera provides a number of tools and dialogs that can all be opened on the screen at the same time. The basic Chimera window provides the main interactive workspace for displaying and manipulating molecular (or volumetric) data. The default Chimera graphics window is pretty small, so if you like, resize the main Chimera window by placing the cursor on any corner and dragging with the left mouse button.
Now open a structure. Choose the menu item File→Open if the files are local. From the resulting dialog, choose and open the previously downloaded file 1zik.pdb (the File type should be set to PDB). If you want to fetch directly from the PDB insted, choose File→Fetch by ID, and type 1zik in the PDB ID field. The structure will appear in the main graphics window. The structure is a leucine zipper formed by two peptides.
Side View showing 1zik
Within the Side View, try moving the eye position (the small square; scales the view) and the clipping plane positions (vertical lines) with the left mouse button. The Side View will renormalize itself after movements, so that the eye or clipping plane positions may appear to "bounce back" after you have adjusted them; however, your adjustments have been applied to the main display.
| Menu Item | Description |
| Color | Colors selected objects. Color target can be limited to object types indicated by the radio buttons |
| Label | Labels selected atoms. residue submenu provides options for residue labeling |
| Focus | Focuses the view on the selected atom(s), zooming and translating if necessary. |
| Atoms/Bonds | Controls the display and representation of atoms and bonds. |
| Ribbons | Controls the display and representation of secondary structure using "ribbons". |
| Surface | Controls the display and representation of molecular surfaces. |
| Target | Restricts target of the action to certain selected or unselected object types. |
| Inspect | Launches the Selection Inspector. |
| Write | Writes the currently selected objects to a file. |
You can now use some menu commands to simplify the display. In general, Chimera Actions act on the current Selection. If nothing is explicitly selected (see Selection section below), then actions will act on everything. The Actions menu can now be used on our model. Explore the Actions menu to see what kinds of functions it provides. As you can see, the Actions menu provides access to some general actions such as Color and Label and some actions that are specific to the type of the underlying representation such as Atoms/Bonds and Surface. The table at the right describes these menu items in more detail. We will use some of these commands later in the tutorial.
To display only the main chain, use the two actions: Actions→Atoms/Bonds→hide to undisplay the model followed by Actions→Atoms/Bonds→chain trace only to display only the chain trace, which shows all of the α-carbons (CA atoms in Chimera) and connects them in the same way that the residues are connected.
| Mouse button | Modifier | Action |
| Btn1 (left button) | |
Rotation |
| Btn2 (middle button) | |
XY Translation |
| Btn3 (right button) | |
Scaling |
| Btn1 | Ctrl | Selection |
| Btn1 | Ctrl-Shift | Add to (or remove from) Selection |
| Btn2 | Ctrl | Z Translation |
Now use the mouse to manipulate the model in the main window.
By
default, the left mouse button controls rotations, the middle mouse
button controls XY translation, and the right mouse button controls
scaling. Note that the Side View
window is reflecting all of the manipulations in the main window.
Continue moving and scaling the structures with the mouse in the
graphics window and Side View
as desired throughout the tutorial.
Next, thicken the lines to
make them more visible (we'll look at other representations later):
Actions→Atoms/Bonds→wire width→3.
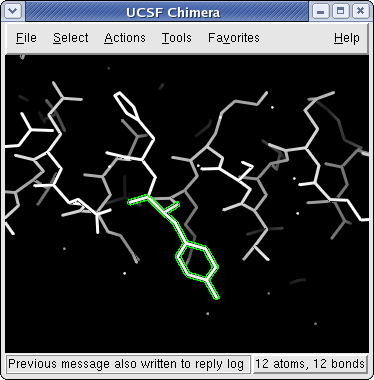
Leucine zipper with Tyrosine 17 (B chain) selected
In addition to using the mouse to select a single atom, use can also use the mouse to drag out a selection area. As before, you depress the Ctrl key and mouse button 1 (usually the left button), and sweep out an area before releasing. All atoms and bonds within that area will be selected. As before, Ctrl-Shift-Btn1 can be used to add to an existing selection, either by clicking or by dragging.
One additional keyboard feature is the use of the ↑ key to increase the scope of a selection. Chimera maintains a hierarchy of objects from atoms to residues to chains to models. If an atom is selected, the ↑ key will select the residue containing that atom. If a residue is selected, the ↑ key will select the chain containing that residue. If a chain is selected, the ↑ key will select the entire model. Similarly, the selection scope can be narrowed by using the ↓ key.
Spend some time selecting various parts of the model. The easiest way to deselect everything is to Ctrl-click in any blank space within the graphics window. Once you are done, reselect the two α-carbons from each of the chains so that we can begin to act on the selections.
1zik Colored by element
Each residue label is of the form:
res_name res_number.chainIt is now evident that one peptide is chain A, and the other is
chain
B. To deselect the atoms, pick in a region of the graphics window away
from any atoms or use the menu item
Select→Clear Selection.
Undisplay the residue labels:
Actions→Label→residue→off
Repeat the process to color chain B yellow. Another way to select an entire chain is to pick an atom or bond in the chain and then hit the up arrow key twice, once to expand the selection to the entire residue and another time to expand it to the entire chain.
There is actually another "chain" in this model, not currently displayed: water. This chain ID was assigned automatically when the structure was read in.
Select→Chain→water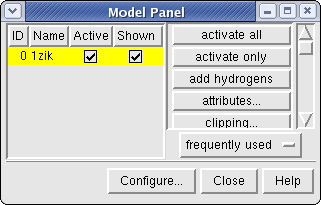
Chimera Model Panel
Chimera provides a number of other tools to observe the state of
the model. One is the Model Panel (
Tools→Inspectors→Model Panel).
A checkbox in the Active column of the Model Panel shows
that the model is activated for motion; unchecking the box makes it
impossible to move. Checking the box again restores the movable state.
Make sure 1zik.pdb is highlighted on the left side of the
Model Panel
(if not, click on it) and then click close in the list of functions on
the right side. Use the Close button at the bottom to close the
Model
Panel. Go on to Part 2 below, or terminate the Chimera session. A
Chimera session may be ended using File→Quit.
6bna.pdb Stick Representation
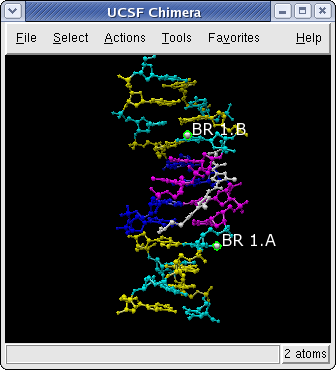
With Chimera started and the Side View opened as described at the beginning of Part 1, choose the menu item File→Open. From the resulting dialog, choose and open the previously downloaded file 6bna.pdb (the File type should be set to PDB). It contains the molecule netropsin bound to double-helical DNA.
Color the different nucleotides different colors.
For example, color
the adenosine residues (adenine nucleotides) blue:
Select→Residue→name→A
Actions→Color→blue
Analogously, color cytosine nucleotides (C residues) cyan, guanine nucleotides (G residues) yellow, and thymine nucleotides (T residues) magenta. Clear the selection by using Select→Clear Selection or picking in a region of the graphics window away from any atoms.
Rotate, translate, and scale the structure as needed to get a better look (see mouse manipulation to review how this is done). Continue moving and scaling the structure as desired throughout the tutorial. There are still many white atoms, including the netropsin molecule in the minor groove of the DNA and water. Undisplay the water:
| Menu Item | Description |
| Action→Atoms/Bonds→wire width | Sets the width (thickness) of the lines in the wire representation |
| Action→Atoms/Bonds→wire | Bonds are displayed as connecting lines, atoms are displayed as dots |
| Action→Atoms/Bonds→stick | Similar to wire, bonds are represented as three-dimentional sticks and atoms are displayed as end-caps |
| Action→Atoms/Bonds→ball & stick | Atoms are displayed as small spheres, bonds are displayed as in stick |
| Action→Atoms/Bonds→sphere | Atoms are displayed as van der Waals spheres |
| Action→Ribbon→show | Display the secondary structure as a "ribbon" |
| Action→Ribbon→hide | Hide the "ribbon" display |
| Action→Ribbon→flat | Use a flat representation for the ribbon |
| Action→Ribbon→edged | Display the ribbon with flag edges |
| Action→Ribbon→round | Display the ribbon with rounded edges |
| Action→Surface→show | Display a molecular surface |
| Action→Surface→hide | Hide a molecular surface |
| Action→Surface→solid | Display a solid surface |
| Action→Surface→mesh | Display the surface as a mesh |
| Action→Surface→dot | Display the surface as dots |
| Action→Surface→transparency | Set the transparency of the surface |
Next, try some different molecular representations. They can be translated, rotated, and scaled interactively. Multiple representation types can be combined with each other and with surfaces (more on surfaces below).
Select→Clear Selection (to ensure that nothing is selected; otherwise, the water is still selected, even though it is invisible)Remember that when nothing is selected, the Actions menu applies to everything.
Actions→Ribbon→show
DNA Helix with bound Netropsin (6bna.pdb)
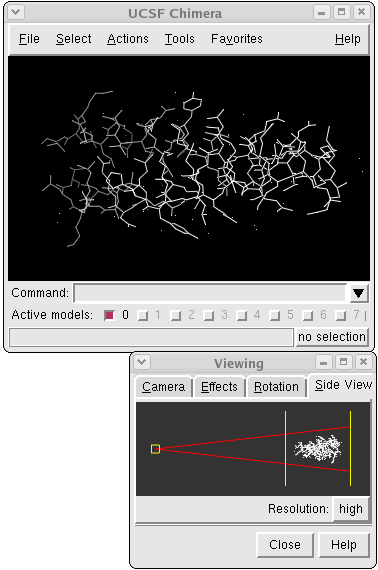
Chimera with Command Tool and Side View
On Linux run the executable "chimera" in the bin directory of your Chimera installation. If Chimera is installed in /usr/local/chimera, then you run /usr/local/chimera/bin/chimera from a shell.
On Windows start Chimera by double clicking the Chimera icon in the directory called bin in your Chimera installation. If Chimera is installed in \Program Files then the executable will be found in directory \Program Files\Chimera\bin. A Chimera icon will also be put on your desktop by default.
On Mac start the Apple X server found in /Applications/Utilities/X11. Then double click the Chimera application to start. The X server is not installed in Mac OS 10.3 by default and if you do not have it you can download it from Apple. It is necessary for running Chimera on the Mac.
Commands are entered into the Command Line and scaling and clipping operations can be performed with the Side View. There are several ways to start each of these tools; one is to choose Tools→Keyboard→Command Line and Tools→Viewing Parameters→Side View from the menu.
Now open the previously downloaded structure:
Command: open 1zik.pdbIf the file is not in the working directory, use File→Open instead, as shown in Part 2. The structure will appear in the main graphics window, and a tiny version is shown in the Side View. The structure is a leucine zipper formed by two peptides. Within the Side View, try moving the eye position (the small square; scales the view) and the clipping plane positions (vertical lines) with the left mouse button. The Side View will renormalize itself after movements, so that the eye or clipping plane positions may appear to "bounce back" after you have adjusted them; however, your adjustments have been applied to the main display.
You can now use some menu commands to simplify the display. In general, a Chimera command consists of the command, one or more arguments, and an optional target. If no target is specified, the command operates on the entire model. This is different from the Menu interface, in which Actions operate on the current Selection. One of the built-in targets provided in the Chimera Command Tool is selected, sel, or picked which applies the command to the current selection.
To simplify the display:
Command: chain @caThis command shows only the atoms named CA (α-carbons) and connects them in the same way that the residues are connected. Try manipulating the structures in the main graphics window with the mouse. By default, the left mouse button controls rotation and the middle mouse button controls XY translation. Continue moving and scaling the structures with the mouse in the graphics window and the Side View as desired throughout the tutorial. Next, thicken the lines:
Command: linewidth 3
In combination with the Ctrl key, the mouse buttons have additional functions. By default, picking from the screen (a type of selection) is done by clicking on the atom or bond of interest with the left mouse button while holding down the Ctrl key. To add to an existing selection, also hold down the Shift key. The selection is highlighted in green. Try picking two α-carbons, one from each peptide (Ctrl-Shift-Btn1). Remember that the Shift key is needed to select both atoms; otherwise, only the most recent selection will be retained.
Label the atoms you have selected:
Command: label selThe label command shows atom information (atom name, by default). Undisplay the atom labels, then show labels for the residues containing the selected atoms:
Command: ~labelEach residue label is of the form:
Command: rlabel sel
res_name res_number.chainIt is now evident that one peptide is chain A, and the other is chain B. To deselect the atoms, pick in a region of the graphics window away from any atoms or use the menu item Select→Clear Selection. Undisplay the residue labels:
Command: ~rlabel
| Symbol | Function | Usage |
| # | model number | # model (integer) |
| #. | submodel number | #.submodel (integer) |
| : | residue | : residue (name or number) |
| :: | residue name | ::residue (integer) |
| :. | chain ID | :.chain |
| @ | atom name | @atom |
| @. | alternate location ID | @.alt_loc |
| - | range | specified a range of models, submodels, or residues |
| , | name separator | separates models or residues, ranges of models or residues, or names of atoms |
| * | whole wildcard | matches whole atom or residue names, e.g., :*@CA specifies the α-carbons of all residues |
| = | partial wildcard | matches partial atom or ressidue names, e.g., @C= specifies all atoms with names beginning with C |
| ? | single-character wildcard | used for atom and residue names only, e.g., :G?? selects all residues with three-letter names beginning with G |
| ; | command separator | separates multiple commands on a single line |
| z< | zone specifier | z<zone or zr<zone spcifies all residues within zone angstroms of the indicated atoms, and za<zone specifies all atoms (rather than entire residues) within zone angstromgs of the indicated atoms. Using > instead of < gives the complement. |
| & | intersection | intersection of specified sets |
| | | union | union of specified sets |
| ~ | negation | negation of specified set (when space-delimited) |
Command: color cyan :.aThere is actually another "chain" in this model, not currently displayed: water. This chain ID was assigned automatically when the structure was read in.
Command: color yellow :.b
Command: disp :.waterdisplays the water (only the oxygens are visible in the X-ray structure);
Command: show :.agets rid of everything except the A chain, but displays all of its atoms;
Command: chain :.a@n,ca,cshows the backbone of the A chain only. If the chain specification ":.a" had been omitted, then the backbones of both chains would have been displayed.
Command: dispdisplays all the atoms and colors them according to element.
Command: color byelement
The Command Line also shows which models are activated for motion: below the command line, 0 should be in bold and the small box next to it highlighted. Clicking the box turns off the highlighting and makes it impossible to move the molecule in model 0. Clicking the box again restores the movable state.
Command: close 0closes the model. Go on to Part 2 below, OR terminate the Chimera session. A Chimera session may be ended using the following command:
Command: stop
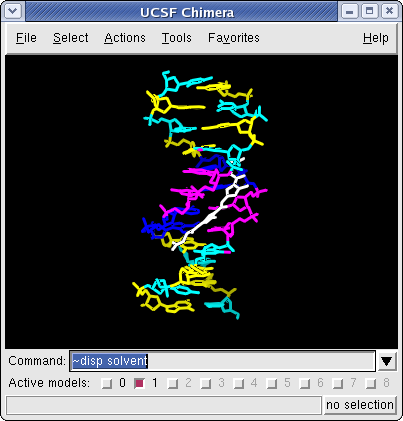
Chimera with Command Tool Showing Netropsin bound to DNA (6bna.pdb)
With Chimera started and the Command Line and Side View opened as described at the beginning of Part 1, choose the menu item File→Open. From the resulting dialog, choose and open the previously downloaded file 6bna.pdb (the File type should be set to PDB). It contains the molecule netropsin bound to double-helical DNA. (Files can also be opened from the Command Line, as shown in Part 1.)
Color the different nucleotides different colors, specifying them by residue name:
Command: color blue :aRotate, translate, and scale the structure as needed to get a better look (see mouse manipulation to review how this is done). Continue moving and scaling the structure as desired throughout the tutorial. There are still many white atoms, including the netropsin molecule in the minor groove of the DNA and water. Undisplay the water:
Command: color magenta :t
Command: color yellow :g
Command: color cyan :c
Command: ~disp :.water
-OR- (these are equivalent)
Command: ~disp solvent
Next, try some different molecular representations. They can be translated, rotated, and scaled interactively. Multiple representation types can be combined with each other and with surfaces (more on surfaces below).
Command: ribbonThe latter command changes only chain B to the stick representation, with the rest remaining in the sphere representation.
Command: ~ribbon
Command: represent stick
Command: repr sphere
Command: rep stick :.b
Note that commands (but not their keyword arguments) can be truncated to unique identifiers. For example, the command represent can be shortened to repr or rep but not re (because other commands also start with re), whereas the keywords stick, sphere, etc. cannot be truncated.
Next, change everything to a ball-and-stick representation:
Command: repr bsIn this representation, pick one of the atoms in the white netropsin molecule. Label the residue,
Command: rlabel pickedshowing that it is named NT and is part of chain HET (assigned automatically when the structure was read in). The residue label may not be very close to the selected atom. Remove the residue label:
Command: ~rlabelTwo white atoms that are not part of netropsin are displayed. They are apparently attached to cytosines, which have been colored cyan (above). Pick the two atoms and label their residues,
Command: rla pickedshowing that one DNA strand is chain A, the other strand is chain B, and each strand contains a brominated cytosine. Use Select→Clear Selection to deselect the atoms, then undisplay the residue labels:
Command: ~rla
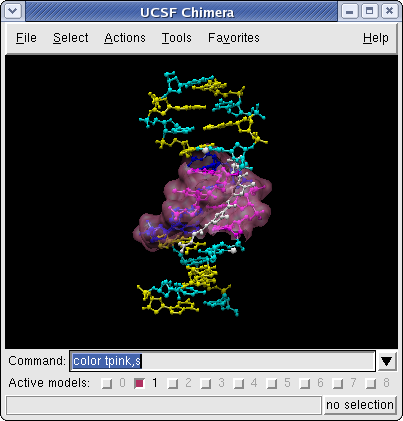
Chimera Surfaces on Netropsin bound to DNA (6bna.pdb)
Finally, have some fun with the surface command. There are built-in categories within structures such as main and ligand; when nothing is specified, surface shows the surface of main. Surfaces can be translated, rotated, and scaled interactively.
Command: surfaceBy default, a surface has the same color as the corresponding atoms; however, surface color can be specified separately:
Command: ~surface
Command: surface ligand-OR- (these are all equivalent)Command: surfrepr mesh
Command: surface :nt
-OR-
Command: surface ::nt
-OR-
Command: surface :.het
Command: surf :g,c
Command: ~surf
Command: surf :a.b,t.b
Command: surf :a,t
Command: surf ligand
Command: color red,s ligandSometimes it is helpful to make a solid surface transparent. One way to do this is to define a transparent color and then use the new color in a command:
Command: ~surf ligand
Command: color green,s :t
Command: surfrepr solid
Command: colordef tpink 1. .5 .7 .4The numbers in the colordef command refer to red, green, blue, and opacity components, respectively.
Command: color tpink,s
Use the command stop to terminate the Chimera session.