

 home
overview
research
resources
outreach & training
outreach & training
visitors center
visitors center
search
search
home
overview
research
resources
outreach & training
outreach & training
visitors center
visitors center
search
search
home
overview
research
resources
outreach & training
outreach & training
visitors center
visitors center
search
search
home
overview
research
resources
outreach & training
outreach & training
visitors center
visitors center
search
search

Tom Goddard¹, Bridget Carragher², and Clint Potter²
¹
Resource for Biocomputing, Visualization, and Informatics
University of California, San Francisco
²
National Resource for Automated Molecular Microscopy
Scipps Research Institute
The National Resource for Automated Molecular Microscopy (NRAMM) was established in December 2002 with the mission of developing, testing and applying technology aimed at automating the processes involved in solving macromolecular structures using cryo-electron microscopy (cryoEM). The goal of automation is not only to facilitate the process of molecular microscopy, although this is a welcome benefit, but also to expand the scope of accessible problems and push experimental frontiers by making possible investigations that are deemed too difficult or high risk because of the considerable effort involved in using manual methods. An additional goal of the automation is to enable much higher data throughputs; this goal is driven by the need to improve resolution for single particle reconstructions by increasing the numbers of particles contributing to the average 3D map. The need for higher throughput is also driven by a variety of biological projects with the need to image conformationally variable structures or very large structures, which result in a low yield of particles per image. The other major mission of NRAMM is to use the infrastructure developed to open up the sometimes esoteric practices of cryoEM to a much wider group of researchers including those from the cell biological, X-ray crystallography, chemical engineering and material science communities.
The final goal of many of the projects supported by NRAMM is a three-dimensional (3D) electron density map of a large molecular machine. Over the past several years Chimera has increasingly been used as the method of choice at NRAMM for examining and analyzing these maps. New users find it very easy to get started with this application so that they can immediately begin examining maps with virtually no training. More experienced users make excellent use of the wealth of specialized tools enabling segmentation, fitting, coarse modeling, measuring and coloring of density maps. These tools are well suited to the examination of large macromolecular structures with resolutions in the range of 0.5-10nm. All of the tools are simple to operate, robust and very interactive, requiring computations of only a few seconds, and are supported by very extensive documentation. A few examples from NRAMM projects where these tools have been used extensively are shown below.
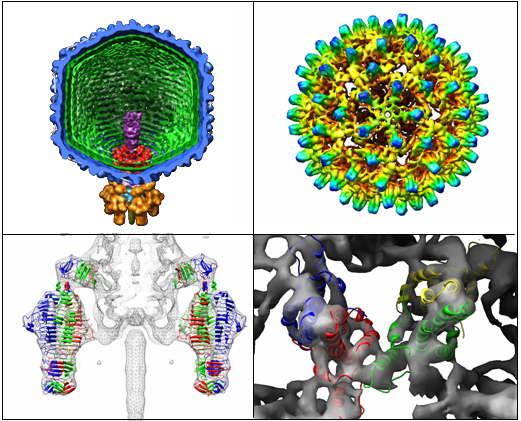
Figure caption: Visualizations of large macromolecular structures using Chimera. The two left hand panels show P22 (Lander, 2006), a large asymmetric bacteriophage and the right panels show the Hepatitus B virus.
One of the major goals of the NRAMM Automated Processing Pipeline (Appion) is to allow us to integrate a variety of routines, algorithms and packages into a single workflow. Once the infrastructure is in place it will provide the flexibility to incorporate new procedures or algorithms into the pipeline with very little overhead of resources and personnel. The key to managing the pipeline, communicating between the disparate routines, and understanding the outputs will be an underlying and tightly integrated database (Fellmann et al., 2002) and a variety of web based tools for querying and viewing the information stored in the database. Over the past few years we have demonstrated the power of this approach by developing a series of databases and viewing tools that integrates the images acquired by our automated data acquisition system, Leginon (Suloway et al., 2005), with analysis and processing routines for contrast transfer function (CTF) estimation and particle selection. This has made it possible to develop complex queries on the parameters and outputs of these routines in order to explore what factors influence the final quality and resolution of the reconstructed maps (Stagg and al., 2006). This required an infrastructure to support insert and query functions to the database by a very wide variety of applications, including Leginon (written primarily in python), ACE (a Matlab extension) (Mallick et al., 2005) and Selexon (a mixture of C codes and Tcl shell scripts) (Zhu et al., 2004).
We plan to incorporate volume viewing capabilities into the Appion pipeline by linking it directly to the UCSF Chimera package. When a user selects a volume to view from the Appion web page, Chimera will be launched automatically on the user's local computer. NRAMM and the UCSF Resource in Biocomputing, Visualization, and Informatics will collaborate to develop appropriate protocols to allow data and parameters to be transferred from the Appion database to Chimera. XML will be used to translate between the parameters generated by the SQL queries to the database and the startup and command files required by Chimera. Much of this infrastructure is almost in place. A more substantial effort will be required to return data and parameters from Chimera back to the database. This capability is critical to the overall philosophy of the pipeline as it will allow users to keep track of complex Chimera environment variables and settings between different experiments and also provide the capability of using the database to keep track of images generated from Chimera and saved to disk. These images will then be available for browsing and viewing by the user. We will also develop capabilities to allow users to apply the Chimera settings used in one experiment to a different experiment to facilitate the comparisons between complex volume data or between volumes generated using different parameters for acquisition or processing.
Additional capabilities within Chimera and the Volume Viewer extension that would benefit this project include:
- Ability to visualize very large volumes (eg. 5123). Perhaps this could be done by scaling down the overall volume but then letting the user look at small parts of it at full resolution.
- We would love to be able to generate some kind of "quantitative" assessment of the reconstructed volumes. That is, we would like to be able to compare one volume to another and also get some measure of the goodness of fit of the volume to the PDB coordinates. We'd like this for both whole volumes as well as segmented out regions.
- We would love to be able to orient the Chimera volume into an orientation determined by the infamous "euler" angles used by the single particle community to describe the orientation of each particle relative to the volume. These are defined differently in every community but our database can be used to translate and there are other tools also being developed to allow for these translations.
References:
- Fellmann, D., Pulokas, J., Milligan, R. A., Carragher, B., and Potter, C. S. (2002). A relational database for cryoEM: experience at one year and 50 000 images. J Struct Biol 137, 273-282.
- Lander, G. C., Liang Tang, Sherwood R. Casjens, Eddie B. Gilcrease, Peter Prevelige, Anton Poliakov, Clinton S. Potter, Bridget Carragher, and Johnson, J. E. (2006). A protein sensor for head full viral chromosome packaging is activated by spooled dsDNA. Science. In Press.
- Mallick, S. P., Carragher, B., Potter, C. S., and Kriegman, D. J. (2005). ACE: automated CTF estimation. Ultramicroscopy 104, 8-29.
- Stagg, S., and al., e. (2006). Automated CryoEM DataAcquisition and Analysis of 284,742 Particles of GroEL. J Struct Biol In press.
- Suloway, C., Pulokas, J., Fellmann, D., Cheng, A., Guerra, F., Quispe, J., Stagg, S., Potter, C. S., and Carragher, B. (2005). Automated molecular microscopy: the new Leginon system. J Struct Biol 151, 41-60.
- Zhu, Y., Carragher, B., and Potter, C. S. (2004). Contaminant detection: improving template matching based particle selection for cryo-electron microscopy. 1071-1074.
Laboratory Overview | Research | Outreach & Training | Available Resources | Visitors Center | Search