

 home
overview
research
resources
outreach & training
outreach & training
visitors center
visitors center
search
search
home
overview
research
resources
outreach & training
outreach & training
visitors center
visitors center
search
search
home
overview
research
resources
outreach & training
outreach & training
visitors center
visitors center
search
search
home
overview
research
resources
outreach & training
outreach & training
visitors center
visitors center
search
search

Jacquelyn S. Fetrow¹, and Patricia C. Babbitt²
¹ Wake Forest University
²
Resource for Biocomputing, Visualization, and Informatics
University of California, San Francisco
Introduction
The Structure-Function Linkage Database (SFLD) is a major resource for structural and mechanistic analysis of functional sites. Integration of additional tools that implement orthogonal approaches for structural analyses of functional sites will provide a better understanding of both the mechanism and evolution of protein function. As described in the core project for the SFLD, mechanistically diverse enzyme superfamilies serve as a good model system for development of sophisticated tools to address previously intractable problems in computational functional inference. For similar reasons, these superfamilies also serve as good models for comparison of structural analysis methods because we can identify commonalities and differences across the family, superfamily and suprafamily members identified by the various methods. Such comparisons will improve our understanding of mechanism and evolution of protein function. In addition, inclusion of these methods into the SFLD will enhance this well curated resource, providing the scientific community with high confidence annotation information, complemented by additional tools for annotating new sequences they identify as members of these superfamilies.Background
Jacquelyn S. Fetrow, Reynolds Professor of Computational Biophysics at Wake Forest University, was an originator of methods that used structural motifs to identify protein functional sites (Fetrow & Skolnick, 1998) and to further develop those motifs so that they could be applied to screening protein sequences (Fetrow & Godzik, 1998). These structural motifs, termed Fuzzy Functional Forms or FFFs, are based on a small set of distances between alpha carbons of "key residues" at the protein active site (see figure below). These motifs have been used to identify potential (Fetrow, 1999) and confirmed (Betz, et. al., unpublished results) functional sites where sequence analysis was unsuccessful. The FFF technology was eventually licensed to a company which created a library of motifs. However, these were not made available to the general research community.In its initial implementation, the FFF technology exhibited two limitations: 1) it focused on a small number of residues (three) at the active site, so identified sites were highly general; and 2) it only provided a "yes" or "no" answer -- no score of confidence for evaluating the results of function identification was developed. Thus, we developed an extension of the FFF method called active site profiling (Cammer, 2003). This method combines sequence and structural information to create an active site signature for the functional site region of a given protein. Signatures are then combined to create an active site profile (ASP), and an empirically derived scoring function indicates relationships between signatures. This method does not rely on residue conservation across an entire family and it is applicable to all functional sites, including enzyme-active sites, regulatory sites, and cofactor-binding sites. Comparison to experimental determination of serine hydrolases indicates that the current version of active site profiling significantly increases the rate of true positive identification and decreases the rate of false positive identification over most state-of-the-art fold-recognition algorithms (Baxter, 2004).
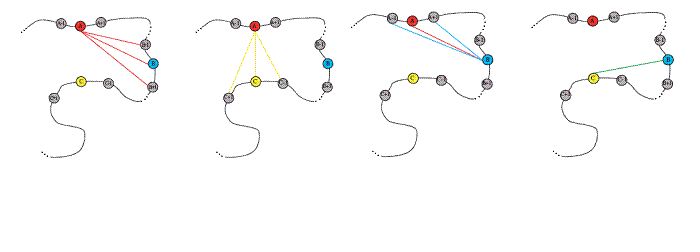
Figure caption: The alpha carbons of the three "key residues" used for FFF construction are shown in red, blue, and yellow. Alpha carbons for residues on either side of the key residue are shown as gray circles. The nine distances that are used to construct the FFF motif are indicated by colored lines between the spheres.
As a major step in enhancing both access of the scientific community to our methods and the utility of the SFLD we propose to collaborate with the RBVI resource to integrate our methods and data into the SFLD. Development of the methods themselves is a funded project (see next section), but requires further work to implement integration with the SFLD. Researchers will benefit from these enhanced RBVI resources and the products will benefit both the RBVI mission and that of our own laboratory.
Ongoing method development
The ongoing development of active site profiling combines computational sequence, structure, bioinformatics and biophysical methods to characterize the molecular function sites of six superfamilies (in particular, six of the superfamilies that are of interest to the Babbitt laboratory, including enolases, crotonases, haloacid dehalogenases, amido hydrolases, and peroxiredoxins). As part of a funded NSF project in my laboratory (MCB-0517343), these superfamilies are classified by enhanced active site profiling methods based on specific biophysical features and evaluated in part by comparison to known biological features and mechanisms. This funded project aims to characterize electrostatics, structure, and sequence features of each functional site, which, we hypothesize, should correlate with mechanism better than current clustering approaches based on global sequence alignment. The specific objectives of this funded project are:
- Characterize the functional site features and use them to develop methods for clustering the Prx family
- Analyze the electrostatics at the Prx functional sites
- Integrate the electrostatic, sequence, and structural information to create a robust profiling method and use it to identify peroxiredoxin subfamilies
- Use these methods to create active site signatures and profiles for the Babbitt lab's "gold standard" protein superfamilies (Brown, 2006).
The long-term goal of this project is to develop an integrated method for describing functional sites that uniquely combines physics, chemistry, and structural information and to use these methods to characterize the general principles that underlie biological mechanism.
In addition, we have funding from the NIH (NCI R21 CA112145) to develop active site profiles for oxidized cysteine post-translational modification sites. Although post-translational modifications are not within the current purview of SFLD, additional ASP technology developed through this funding source will also be available for integration into the SFLD.
We have also begun to re-develop the FFF library and technology within the academic domain. Although not yet funded (we are developing significant preliminary data; E. E. Pryor, Computer Science Masters Thesis, May 2006), methods and technologies developed will also be made available for integration into SFLD.
These projects will create a minimal web site that will make the methods available to the community. We have already begun construction of that site called DASP ("Deacon Active Site Profiler;" www.deac.wfu.edu (Fetrow, 2006); however, funding does not allow us to create a fully integrated and enhanced resource. Significant value will be realized if our methods and those from the Babbitt lab are integrated and the output combined. Integration with the SFLD will provide such a fully integrated and enhanced resource that will be of value to a much larger user community.
Integration with SFLD
To integrate ASP and FFF technology into the SFLD, we propose to develop the tools and user interfaces that will provide:
- Integration of the DASP web site with the SFLD, which will allow researchers to build active site profiles and to search sequence databases with those profiles (a nascent version of this is currently available at the DASP web site);
- The ability for users to determine whether their protein(s) match a protein family by allowing them to compare their sequence(s) to the active-site profiles for the families and superfamilies that are represented in the SFLD;
- Classification of known structures based on FFF and ASP analysis, of the families, subgroups, superfamilies in the SFLD;
- Comparisons between the FFFs, the active site profiles (and clusters of those profiles) and the SFLD clusters.
The implementation of the FFF technology in collaboration with the SFLD will allow researchers to take subsets of the residues identified as critical for enzyme function and identify similar motifs in other proteins of known structure. Integration of the ASP technology, and implementation of tools for clustering the active site profiles, will allow researchers to identify the similarities and differences between the active sites of the members of these superfamilies.
References:
- Baxter, S. M., J. S. Rosenblum, et al. (2004). "Synergistic computational and experimental proteomics approaches for more accurate detection of active serine hydrolases in yeast." Mol Cell Proteomics 3: 209- 225.
- Brown, S.D., Gerlt, J.A., Seffernick, J.L., and Babbitt, P.C. (2006). "A gold standard set of mechanistically diverse enzyme superfamilies." Genome Biol 7:1-15.
- Cammer, S. A., B. T. Hoffman, et al. (2003). "Structure-based active site profiles for genome analysis and functional family subclassification." J Mol Biol 334(3): 387-401.
- Fetrow, J. S., A. Godzik, et al. (1998). "Functional analysis of the Escherichia coli genome using the sequenceto- structure-to-function paradigm: identification of proteins exhibiting the glutaredoxin/thioredoxin disulfide oxidoreductase activity." J Mol Biol 282(4): 703-11.
- Fetrow, J. S. and J. Skolnick (1998). "Method for prediction of protein function from sequence using the sequence-to-structure-to-function paradigm with application to glutaredoxins/thioredoxins and T1 ribonucleases." J Mol Biol 281(5): 949-68.
- Fetrow, J. S., N. Siew, et al. (1999). "Structure-based functional motif identifies a potential disulfide oxidoreductase active site in the serine/threonine protein phosphatase-1 subfamily." Faseb J 13(13): 1866- 74.
- Fetrow, J. S. (2006). "Active site profiling to identify protein functional sites in sequences and structures." Curr Prot Bioinform In press.
Laboratory Overview | Research | Outreach & Training | Available Resources | Visitors Center | Search