

 about
projects
people
publications
resources
resources
visit us
visit us
search
search
about
projects
people
publications
resources
resources
visit us
visit us
search
search
Quick Links
Recent Citations
Parental histone transfer caught at the replication fork. Li N, Gao Y et al. Nature. 2024 Mar 28;627(8005):890–897.
Structure and function of the Arabidopsis ABC transporter ABCB19 in brassinosteroid export. Ying W, Wang Y et al. Science. 2024 Mar 22;383(6689):eadj4591.
Flexibility of binding site is essential to the Ca(2+) selectivity in EF-hand calcium-binding proteins. Lai R, Li G, Cui Q. J Am Chem Soc. 2024 Mar 20;146(11):7628-7639.
Structural dynamics of RAF1-HSP90-CDC37 and HSP90 complexes reveal asymmetric client interactions and key structural elements. Finci LI, Chakrabarti M et al. Commun Biol. 2024 Mar 2;7(1):260.
Activation of human STING by a molecular glue-like compound. Li J, Canham SM et al. Nat Chem Biol. 2024 Mar;20(3):365-372.
Previously featured citations...Chimera Search
Google™ SearchNews
October 30-31, 2023
Planned downtime: The Chimera and ChimeraX websites and associated web services will be unavailable Oct 30 8am PDT – Oct 31 11:59pm PDT.
April 19, 2023
Chimera production release 1.17.1 is now available, fixing an issue with 1.17 for Windows and Linux. See the release notes for details.
April 13, 2023
Chimera production release 1.17 is now available. Updating is required to keep using the tools that run Blast Protein, Modeller, and multiple sequence alignment with Clustal Omega or MUSCLE, as these will soon stop working in older versions. See the release notes for details.
Previous news...Upcoming Events

UCSF Chimera is a program for the interactive visualization and analysis of molecular structures and related data, including density maps, trajectories, and sequence alignments. It is available free of charge for noncommercial use. Commercial users, please see Chimera commercial licensing.
We encourage Chimera users to try ChimeraX for much better performance with large structures, as well as other major advantages and completely new features in addition to nearly all the capabilities of Chimera (details...).
Chimera is no longer under active development. Chimera development was supported by a grant from the National Institutes of Health (P41-GM103311) that ended in 2018.
Feature Highlight

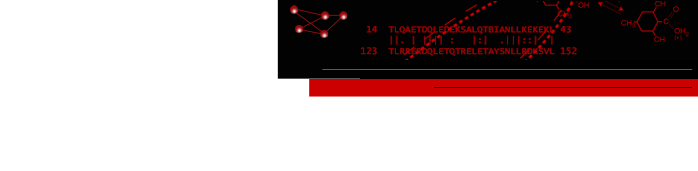
One use of Multidomain Assembler is to set up comparative modeling and concatenation of existing structures to generate a full-length model of a multidomain protein. However, even without model-building, the byproduct is also useful: a visual summary of the structures available for a query sequence, optionally filtered by criteria such as BLAST score and % identity, laid out horizontally in approximate N→C order relative to the query. Overlapping hits are stacked vertically, and segments without structural coverage are indicated with spheres. By default, the multiple sequence alignment of the hits to the query is also displayed.
The figure shows the results of command:
mda p08648 ~/Desktop/MDA limit 4 percent 50
with sequence mismatches in red and molecules other than the hit
chains in blue. Text and pointers have been added with
2D
Labels.
Multidomain Assembler is described in a paper.
(More features...)Gallery Sample
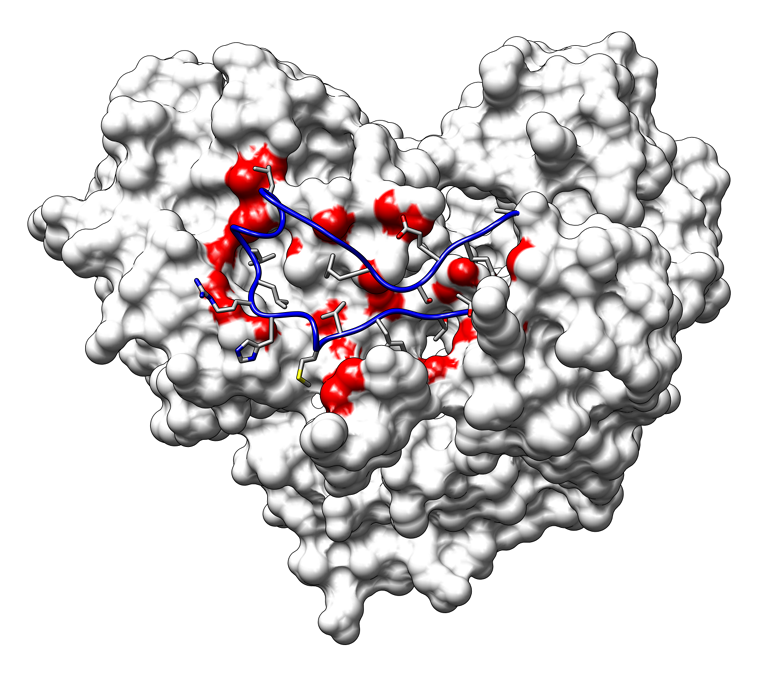
The image shows interactions of the delta-1 loop with the rest of hepatitis C virus RNA-dependent RNA polymerase (Protein Data Bank entry 1quv). Loop residues in contact with the rest of the structure (van der Waals overlap ≥ 0.01 Å) are displayed as sticks; interacting surface atoms are shown as red patches. (More samples...)
About RBVI | Projects | People | Publications | Resources | Visit Us
Copyright 2018 Regents of the University of California. All rights reserved.