
MD Movie is a tool for trajectory (or ensemble) viewing and analysis. For examples of use, see the Trajectory and Ensemble Analysis tutorial. See also: Morph Conformations
There are several ways to start MD Movie, a tool in the MD/Ensemble Analysis category.
Several formats are supported:
| Format | Required Inputs |
|---|---|
| Amber |
|
| CHARMM, NAMD, or X-PLOR |
|
| GROMACS | |
| GROMOS |
|
| MMTK |
|
|
PDB, multiple files
(one file per frame) |
Starting and ending file names must be supplied. The file names must include frame numbers, and files for all intermediate frames must also be present. The files must not contain END records. |
|
PDB, single file
(coordinates for each frame bracketed by MODEL and ENDMDL records) |
The name of the file must be supplied. The MODEL numbers are interpreted as frame numbers and should start with 1 or 0 and increment by 1. |
These input files can be gzipped.
The PDB options generally require coordinates for the same set of atoms to be supplied for each frame. However, if the coordinates supplied for a frame represent only a subset of the atoms in the preceding frame, it will be inferred that the remaining atoms are present but have the same coordinates as in the preceding frame. Changes are cumulated in the forward direction and based on all frames, even when frames are skipped during playback (step size > 1).
All or a contiguous subsegment of a trajectory can be loaded. If pipe is entered as the ending frame number, the input trajectory will be read until it runs out, as long as no other input specifies a smaller ending frame number. For example, for a GROMOS trajectory, the NSTLIM variable in the PROMD file should also be increased to at least the total number of frames expected. When a pipe has been specified, there will be an attempt to load the entire trajectory up front.
A further option is a metafile, simply a text file that specifies the input files/parameters. The first line designates trajectory type (such as amber or gromos, where case is unimportant) and optionally, starting and ending frame numbers of the range to be loaded. A pipe can be specified as described above. If no frame numbers are supplied, the entire trajectory will be loaded; however, if "?" is given for both the starting and ending frame numbers, a dialog for entering this information will appear. The remaining lines specify input files and parameters in the same order as in the dialog. For example:
or:amber 500 1000 dna.prmtop dna.mdcrd
If input files are not in the same directory as the metafile, their pathnames relative to the location of the metafile should be supplied. A metafile can be opened directly from the Command Line (or the system command line upon startup) using the prefix md: or movie:. This will start MD Movie and open the trajectory data.pdb single 108d.pdb
After input files and parameters have been specified, the first set of coordinates will be displayed and the MD Movie controller will appear:
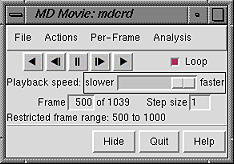
|
If a subsegment was specified, a message about the restricted frame range will be displayed temporarily. The total number of frames in the trajectory (whether loaded or not) will continue to be reported. For all input formats except PDB, the coordinates for a given frame are not read in until that frame is viewed or used for analysis.
From left to right, the buttons mean: play backward continuously; go back one step; stop; go forward one step; and play forward continuously. When the Loop option is on, forward play can wrap from the end to the beginning of the loaded trajectory and reverse play can wrap from the beginning to the end. The rate of continuous play can be adjusted with the Playback speed slider; up to a 1-second delay can be added between frame advances. The Frame number is reported and can also be entered directly to view a specific frame. The Step size controls the level of sampling for viewing purposes. For example, a step size of 3 indicates that only every third frame will be viewed; however, when forward play loops from end to beginning, the movie will start at the first frame loaded, and when reverse play loops from beginning to end, the movie will start at the last frame loaded. Frame number and step size changes take effect when return (Enter) is pressed.
Protein secondary structure assignments are not recomputed automatically over the course of a trajectory. If displaying protein as a ribbon, see the note below.
The current frame or all frames that have been viewed can be saved as a PDB file with File... Save PDB. The view can be held steady on selected atoms. One can define scripts to be executed at each frame update. A complete menu listing with short descriptions is included below.
Hide closes the interface without exiting from MD Movie; the interface can be reopened using the Tools menu entry for the instance of MD Movie. Quit exits from MD Movie and removes the structure from the Chimera window. Help opens this manual page in a browser window.
Images can be captured during trajectory playback and automatically assembled into a movie file. For a one-to-one relationship between trajectory frames and recorded image frames, the Playback speed slider should be moved to the far right. Otherwise, identical image frames will be inserted between the unique data frames to slow playback. The frame number parameters described below refer only to the unique data frames.
Note: Protein secondary structure assignments are not recomputed automatically over the course of a trajectory. If a protein or peptide is displayed as a ribbon and significant conformational changes are occurring, users may want to reassign secondary structure at each frame. This can be done with a per-frame script that includes the Chimera command ksdssp.
File... Record movie opens an interface for specifying image capture and movie assembly parameters.
Close closes the dialog without initiating recording. Help opens this manual page in a browser window.
See also: making movies
Per-Frame... Define script allows specification of a script to be executed at each trajectory frame. Scripts can be written in Chimera commands or Python code, and can incorporate trajectory frame numbers. Python scripts can also access the molecule model instance.
Examples:
chain @ca ~disp solvent hb line 2 color yellow reveal true
from chimera import runCommand
frame = mdInfo['frame']
if frame == mdInfo['startFrame']:
runCommand("roll y 3 40")
elif frame == 10:
runCommand("color green")
elif frame == 50:
runCommand("color orange")
runCommand("rl :ala; color dodger blue :ala; rep stick :ala")
elif frame == 90:
runCommand("~rl; rep wire :ala")
else:
runCommand("color byhet")
Note: Frame arguments in commands such as roll refer to image frames rather than to unique data (trajectory) frames. For a one-to-one correspondence between image and trajectory frames, the Playback speed slider must be positioned all the way to the right and the viewing step size set to 1. Whereas an MD Movie per-frame script executes at each trajectory frame, commands to be executed at each image frame (independent of any trajectory) can be specified with the perframe command.
Insert text file allows browsing to a text file and placing its contents in the script area. Save to file saves the current contents of the script area to a text file.
OK and Apply execute the script with and without closing the dialog, respectively. If the movie is playing, the script will continue to be executed for each trajectory frame until Stop running script is chosen; if the movie is halted on a single frame, the script will be executed for that frame and will not be executed again until a different frame is shown. Clear deletes the contents of the script area. Close closes the dialog without executing the script, and Help opens this manual page in a browser window.
It may be interesting to see which regions of space are highly populated by certain atoms relative to others in the trajectory or ensemble. For example, cations or water hydrogens may tend to occupy space around a negatively charged solute group. Occupancies can be represented as a three-dimensional grid of values, or volume data. Analysis... Calculate occupancy can be used to generate such data and display it with Volume Viewer. The occupancy map can be saved to a file and later reopened in Chimera.
Usually, one should first define a reference set of atoms to hold steady (the trajectory frames will be transformed to keep these atoms in the same place and orientation, as much as possible). This is accomplished by selecting the desired reference atoms and then choosing Actions... Hold selection steady. When the selection is later changed, the "hold steady" atoms will not change unless Actions... Hold selection steady is used again.
Next, one should select the atoms for which occupancy data will be collected. The atoms in the selection can be combined into a single set of occupancy data, or segregated by atom type.
Close closes the dialog without initiating the calculation. Help opens this manual page in a browser window.
For each frame-to-frame comparison, the least-squares-fit RMSD between the indicated sets of atoms will be calculated, without applying any transformation.
OK and Apply initiate the RMSD calculations with and without closing the dialog, respectively. Close closes the dialog without initiating the calculation. Help opens this manual page in a browser window.
The calculations may require additional frames to be read (frames within the loaded range are not actually read until viewed or otherwise used) and may take several minutes, depending on the size of the system and the number of frames. The Abort button on the RMSD map dialog allows termination of a calculation in progress.
The following approaches are recommended for examining long trajectories or long trajectory segments:
Multiple RMSD maps can be open at the same time. However, a given map can be recolored (without recalculation of RMSD values) using the map's menu option RMSD... Change thresholds... to adjust the white/black threshold settings. When the cursor is placed over a map, the corresponding frame numbers and RMSD value are given near the bottom of the dialog. Clicking on a map places the corresponding frame numbers in the Frame fields. Clicking Go changes the graphics display to the corresponding frame.
Alternatively, the Ensemble Match tool can be used to calculate all-by-all pairwise RMSD values for an ensemble read from a single PDB file. The file would need to be opened in a standard way rather than with MD Movie. Unlike MD Movie, Ensemble Match can perform the corresponding pairwise superpositions (i.e. match one structure to another), but its use is impractical for ensembles with very many members.
MD Movie uses the Trajectory module to read the various formats (chimera/share/Trajectory, where chimera is the Chimera installation location). Trajectory contains a subdirectory, formats, which in turn contains subdirectories that each correspond to the Python module for a supported format. By convention, the module (and directory) name for each format is the name of the format with the first letter of each word capitalized and all other letters lowercase. For example, the MMTK module's name is Mmtk.
A format's module is typically structured so that the code that interfaces with Trajectory's generic format handling is in __init__.py, and the code specific to reading a particular format is in another python file, usually named after the format itself (for example, Gromos.py).
__init__.py must support the following:
The format-specific .py file defines an ensemble class that gets instantiated from __init__.py's loadEnsemble function. The ensemble class must support the following methods: