|
|
|
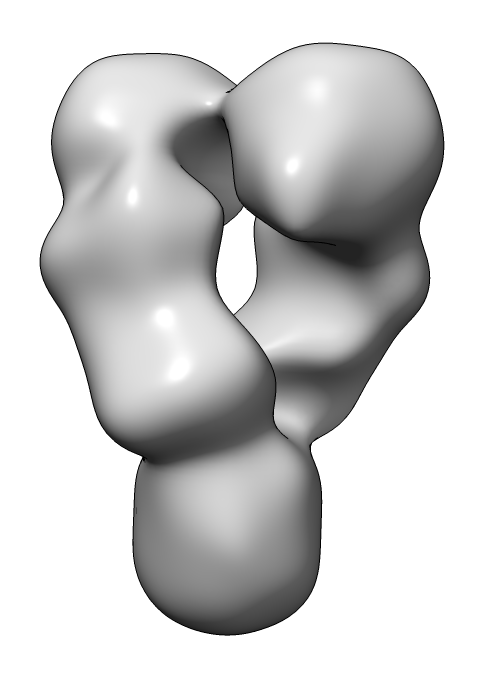
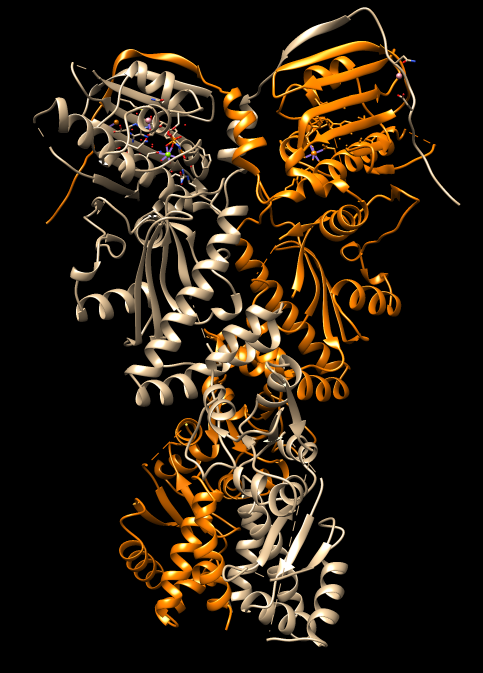
| HSP90 EM map | HSP90 PDB 4ipe |
Tom Goddard
December 11, 2017
UCSF Biophysics 204A tutorial
How to dock an HSP90 atomic model (x-ray) into a low resolution (~20 Angstrom) negative stain electron microscopy single-particle map using UCSF Chimera version 1.12.
| Operation | Chimera menu entry |
|---|---|
| Open HSP90 protein, PDB 4ipe | File / Fetch by ID. |
| Select chain A | Select / Chain / A. |
| Color chain A orange | Actions / Color / Orange. |
| Clear selection | Ctrl-click on background. |
| Operation | Chimera menu entry |
|---|---|
| Open HSP90 map | File / Open.... |
| Adjust map surface threshold | Volume Viewer panel, move vertical bar on histogram. |
| Freeze map motion | Favorites / Model Panel. Click active button (column headed by "A") off for map. |
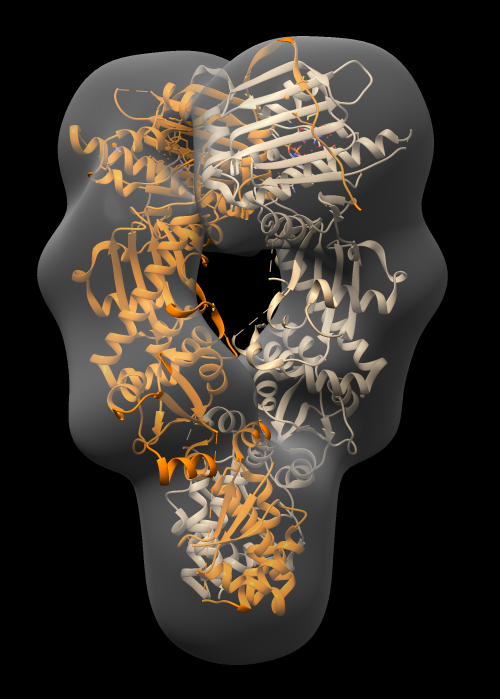
| Hand dock model | Drag atomic model into map with mouse. |
| Unfreeze map | Click active button in Model Panel for map. |
| Optimize fit | Tools / Volume Data / Fit in Map. Press Fit button on Fit in Map panel. |
| Make surface transparent | Press Volume Viewer panel color button, click opacity, change A (opacity) value. |
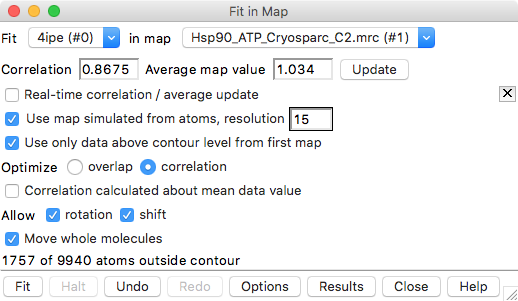
This fit optimization rotated and shifted atomic model to maximize the average map value at the atom positions. Reports number of atoms outside displayed map contour, for example
1754 of 9940 atoms outside contour
| Operation | Chimera menu entry |
|---|---|
| Fit with simulated map | Press Options button on Fit in Map, click Use map simulated from atoms..., set resolution 15 Angstroms |
| Set optimization mode | click Optimize correlation |
| Optimize fit | click Fit. |
| Show simulated map. | In Model Panel check button in S (show) column for molmap 4ipe res 15. |
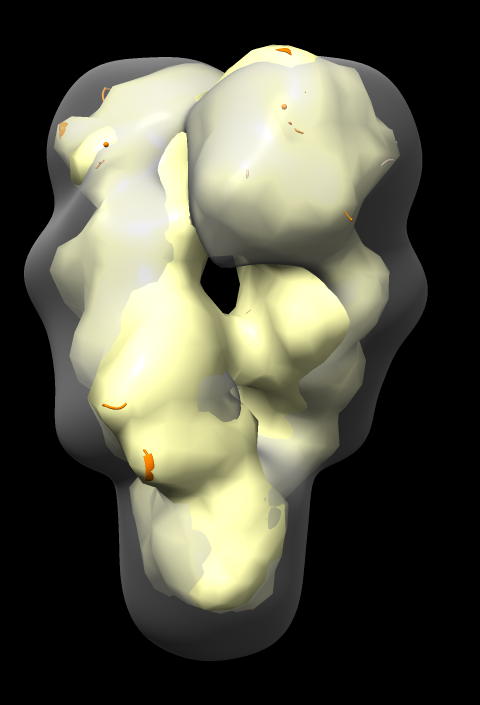
Fit in Map panel reports correlation coefficient and average map value, for example,
Correlation 0.8675 Average map value 1.034
Correlation values compare two maps and range from -1 to 1, with 0 indicating no correspondence, and 1 identical maps.
Notice that atomic model pokes out of map surface in an asymmetric way, the helices and turns of the two chains that poke out do not match. Why?
Is the 4ipe atomic model symmetric?
| Operation | Chimera menu entry |
|---|---|
| Open second copy of 4ipe | Press lightning bolt icon  in lower right corner, click 4ipe button. in lower right corner, click 4ipe button.
|
| Hide maps | unclick shown column buttons for maps in Model Panel |
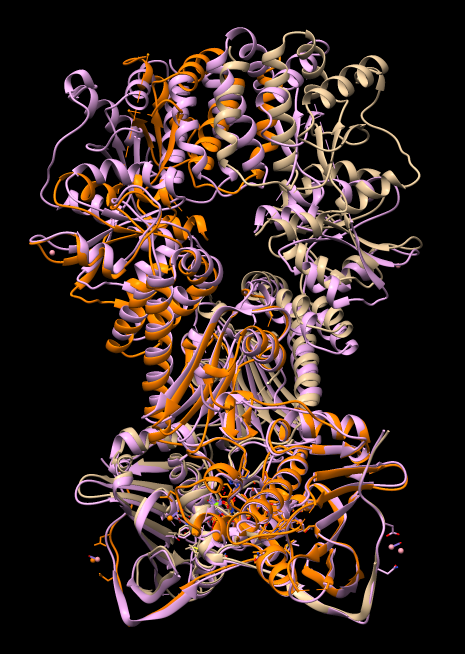
| Align chain B of one 4ipe with chain A of other 4ipe | Tools / Structure Comparison / Matchmaker. Chain pairing: specific chains with specific chains. Reference chain #0 A, match #2 B. Press Apply. |
Note the N-terminal domains align well but the C-terminal domains misalign by more than 10 Angstroms.
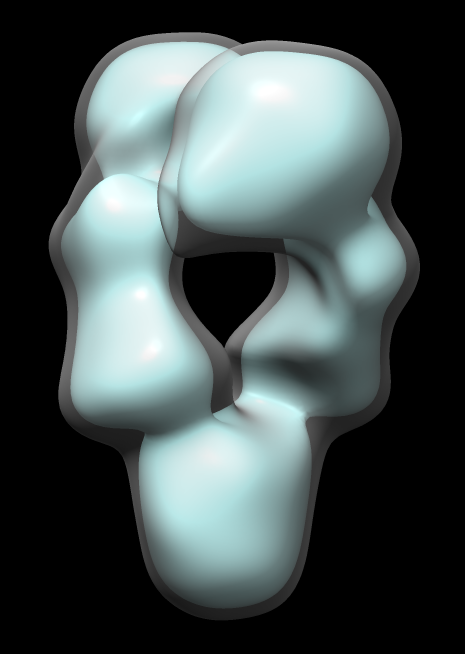
Map single-particle reconstruction imposed C2 symmetry. But let's check that it really has C2 symmetry.
| Operation | Chimera menu entry |
|---|---|
| Open second copy of map | Press lightning bolt icon in lower right corner, click Hsp90_ATP_Cryosparc_C2.mrc button.
|
| Hand align map copy to original map | Click line in Model Panel for map copy, press Activate Only button. Move map copy with mouse to align with original copy. Press Activate All button. |
| Optimize fit of map copy in original map | Tools / Volume Data / Fit in Map. Choose map copy and original map in Fit panel top line. Press Fit |
| Rotate map copy and fit again | Use Model Panel to activate only map copy, approximately hand rotate 180 degrees. Press Fit button. |
Note correlation coefficient is 1 for map fit into map even after 180 degree rotation. Has exact 2-fold (C2) symmetry.
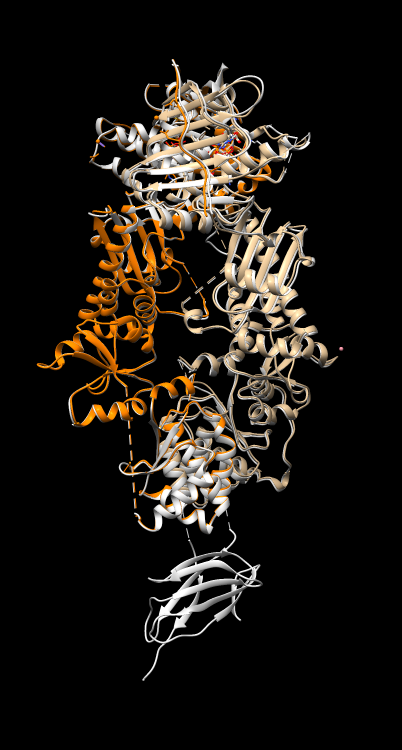
Do other HSP90 atomic dimer models have symmetry? Align another PDB HSP90 model 5tth to 4ipe to see how similar they are.
| Operation | Chimera menu entry |
|---|---|
| Open 5tth | File / Fetch by ID |
| Align 5tth to 4ipe | Tools / Structure Comparison / Matchmaker. Reference 4ipe chain A, match 5tth chain B. Press Apply. |
| Check RMSD difference. | Favorites / Reply Log |
Visually 4ipe is nearly identical to 5tth except the latter has an extra domain. The reply log reports 0.464 Angstroms root mean square deviation (C-alpha atoms) for 587 paired residues.
RMSD between 587 pruned atom pairs is 0.464 angstroms; (across all 588 pairs: 0.471)
Can find 348 other PDB structures with high sequence similarity (BLAST E-value < 1e-38) using menu Tools / Sequence / Blast Protein. Can select some or all and press "Load" to load them and automaticallly align them.