
This tutorial focuses on using the Model Panel and handling ensembles of structures (such as those determined by NMR).
We will view solution structures of a toxin that binds to sodium channels. Separate ensembles were determined for cis- and trans-proline conformations of this toxin:
NMR solution structures of δ-conotoxin EVIA from Conus ermineus that selectively acts on vertebrate neuronal Na+ channels. Volpon L, Lamthanh H, Barbier J, Gilles N, Molgó J, Ménez A, Lancelin JM. J Biol Chem. 2004 May 14;279(20):21356-66.Start Chimera by clicking or doubleclicking the Chimera icon
A splash screen will appear, to be replaced in a few seconds by the main Chimera graphics window or Rapid Access interface (it does not matter which, the following instructions will work with either). If you like, resize the Chimera window by dragging its lower right corner. Show the Command Line by choosing Tools... General Controls... Command Line from the menu.
If you have internet connectivity, structures can be obtained directly from the Protein Data Bank:
Command: open 1g1pIf you do not have internet connectivity, you can download the files 1g1p.pdb.gz and 1g1z.pdb.gz into your working directory and then open them in that order as local files (with File... Open). It is not necessary to uncompress the files.
Command: open 1g1z
Use the ribbons preset:
Menu: Presets... Interactive 1 (ribbons)This may or may not change the appearance, depending on your preference settings.
Rotate, translate, and scale the structures as desired throughout the tutorial. There are several ways to start tools, including from the menu or with a command. Open the Model Panel:
Command: start Model PanelThe Model Panel lists models on the left and functions on the right.
A file of coordinates opened in Chimera becomes a model with an associated ID number and model color. Some PDB files are further subdivided into multiple structures designated with MODEL and ENDMDL records; these are assigned submodel numbers. Since each structure can be handled independently, the general term “model” can refer to a submodel as well as a model that is not subdivided.
Each of these PDB files is an NMR ensemble of several structures (submodels). At first, the submodels in a model are collapsed into a single row. Expand the listing to individual submodels:
|
Repeat the process for the other model. 1g1p (trans-proline conformations, white) has been opened as models 0.1–0.18 and 1g1z (cis-proline conformations, magenta) as models 1.1–1.18.
By default, the list of functions on the right side of the Model Panel includes only those classified as favorites. If you do not see some function that is mentioned, try changing from favorites to all functions using the checkbutton below the list.
Scroll down the list of models in the left side of the Model Panel and choose model 1.2. Try various functions:
show only - hide the other modelsBy default, ribbons suppress backbone atom display. Hide the ribbon to reveal the backbone atoms:
show all atoms - display all atoms
select - select the entire model for further operations
Menu: Actions... Ribbon... hideSelect and delete hydrogens in all models:
Menu: Select... Chemistry... element... HWhen atoms might be needed later, hide should be used instead of delete.
Menu: Actions... Atoms/Bonds... delete
Back to the Model Panel:
sequence... (it may be necessary to change from favorites to all to see this function in the Model Panel) opens a sequence window for the model. Two very short β-strands (positions 24-25 and 29-30) are highlighted in light green. Placing the mouse cursor over a residue in the sequence shows the corresponding structure residue number at the bottom of the sequence window. The β-strand locations were read from the input file along with the coordinates. Highlight a string of residues in the sequence with the mouse and see how they become selected in the structure. Quit from the sequence window, then act on the selection with the menu: Actions... Color... orangeClear the selection (Ctrl-click in an empty area of the graphics window) and go back to showing only ribbons:
Menu: Presets... Interactive 1 (ribbons)Note the orange coloring is gone; interactive presets reset the coloring, whereas publication presets do not adjust colors, aside from making the background white.
The ribbon shows the β-strands as arrows. Although the input file specifies the strand locations as 24-25 and 29-30, the paper describes three β-strands, comprised of residues 8-10, 23-26, and 28-31. Such differences are common because secondary structure assignments are method- and parameter-dependent. Secondary structure assignments could be recomputed with ksdssp, but if the desired assignments are already known, it is much more efficient to change them directly:
Command: setattr r isStrand true :8-10,23-26,28-31This command assigns values of the residue attribute named isStrand. In this case, it is not necessary to set isStrand (or isHelix) to false for any residues, since the original strand residues are still in strands and none of the new strand residues were in α-helices. Secondary structure assignments can also be changed by selecting residues and using the Selection Inspector.
The strands can be emphasized with color:
Command: color blue #1.2(For those who prefer a graphical interface, there is also a Color Secondary Structure tool and associated color by SS function in the Model Panel.)
Command: color yellow #1.2 & strand
In the Model Panel, the S (Shown) checkboxes toggle model display without changing the display settings of individual atoms, bonds, and ribbon segments.
uncheck the S box for model 1.2The modeldisplay command does the same thing. Show all of the models:
check the S box for model 1.2
Command: modeldispThe A (Active) checkboxes in the Model Panel control what can be moved:
uncheck the A box for model 1.2 and try moving the structures with the mouse; now only the other models can be rotated and translatedReset to the original model positions:
check the A box for model 1.2 and try moving the structures again
Command: resetWe will use Ensemble Cluster to cluster each ensemble and identify representative structures, then Ensemble Match to compare the representatives.
Start Ensemble Cluster (under Tools... MD/Ensemble Analysis) and choose 1g1p as the ensemble to cluster. Leave the Parts to Match blank to use all atoms and click OK. Results are shown in a cluster list dialog; three clusters were found. In that dialog,
if options are not shown under Treatment of Chosen Clusters, click the black arrowhead to reveal themNow the submodels of 1g1p (#0) are shown in three different colors for the three clusters, and only the three cluster representatives are chosen in the left side of the Model Panel. In the right side of the Model Panel, click show only to hide all models except those three representatives. Quit from the cluster dialog, then delete the undisplayed members of the 1g1p ensemble:
set the treatment options to Color [all members] and Choose [representatives] in Model Panel
choose all three cluster lines in the dialog with the mouse
Command: delete #0/!displayThis command deletes models with ID number 0 and model display turned off.
Start Ensemble Cluster again and cluster 1g1z using all atoms. This time, four clusters are found. Keeping the treatment settings the same as used before, choose just the two clusters with more than one structure, and again click show only in the Model Panel. Quit from the cluster dialog and delete the undisplayed members of the 1g1z ensemble:
Command: delete #1/!displayIn the left side of the Model Panel, drag with the mouse to choose all five remaining models. Use Model Panel functions to show them, assign them unique colors, and spread them out into a plane:
show - show all five modelsFinally, compare the structures with Ensemble Match (under Tools... MD/Ensemble Analysis). Choose one ensemble as the reference and the other as the alternative. For Parts to Match just specify the backbone atoms:
rainbow... click OK to rainbow-color the models using default settings
tile... click OK to spread the models out into a plane
@n,ca,c,o
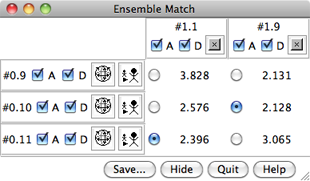 |
Click OK to calculate the matches. Results are shown as a 3 x 2 (or 2 x 3) table with entries for all pairwise comparisons between the ensembles. The A and D buttons control model activation for motion and model display, respectively. The numbers in the table are pairwise RMSDs using the atoms that were specified as Parts to Match.
The structures are not yet superimposed. Clicking a button next to an RMSD value performs the corresponding match and reports in the status line the number of atom pairs used. Superimpose each cis-proline conformation in model 1 on the most similar (lowest-RMSD) trans-proline conformation in model 0.
In this case, cis and trans refer to the peptide bond between leucine-12 and proline-13. Display these residues as ball-and-stick:
Command: disp :12-13The proline rings look somewhat distorted, especially in the cis-proline structures (currently red and yellow). By default, the ribbon path is a smoothed B-spline that does not pass exactly through the true positions of the backbone atoms. Bonds to backbone atoms are drawn to the ribbon and thus may appear to be stretched. Try a cardinal spline instead, which gives a messier-looking ribbon but follows the backbone atom positions more closely, even when some smoothing is applied:
Command: repr bs
Command: ribspline cardinalTo return to the default B-spline ribbon:
Command: ribsp card smooth strand
Command: ribsp bsplineWhen finished viewing the structures, choose File... Quit from the menu to exit from Chimera.