January 12, 2007
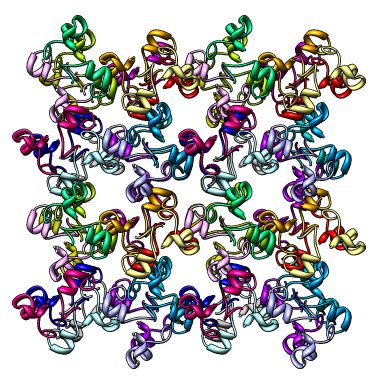
The crystalcoords.py code opens a PDB file and then writes several copies of that PDB model with transformed coordinates to fill out a block of crystallographic unit cells.
The path for the input PDB file and the path prefix for the output PDB files and the number of unit cells along each axis appear at the top of the script and should be edited as needed. Output files are named according to the prefix path with a suffix indicating the unit cell number and the symmetry number, for example PREFIX_110_4.pdb.
This code does not provide a Chimera command or a graphical user interface for writing the coordinates.
The script can be run by opening it with Chimera menu entry
File / Open
or it can be run as a command line argument when Chimera is started from a shell
% chimera crystalcoords.py
or it can be run without the any Chimera window appearing using shell command
% chimera --nogui crystalcoords.py
The script can be in any directory.
The code uses the CRYST1 record from the PDB header to determine unit cell parameters. It reads SMTRY remarks and MTRIX records in the PDB header in order to determine the layout within one unit cell. If SMTRY remarks are missing it uses the space-group listed in the CRYST1 record to determine the correct symmetries.
Most of the routines used by this code are found in the Chimera distribution in the PDBmatrices module:
chimera/share/PDBmatrices/crystal.py
Jan 12, 2007.
Written for Brady Bernard.