
This tutorial focuses on aligning structures and creating sequence alignments with Chimera. The MatchMaker tool is used to align protein structures (create superpositions) and Match -> Align is used to generate a multiple sequence alignment based on the fit of the proteins in space. Sequence alignments are displayed in Multalign Viewer, which is covered in more detail in the Sequences and Structures tutorial.
On Windows/Mac, click the chimera icon; on UNIX, start Chimera from the system prompt:
unix: chimeraA basic Chimera window should appear after a few seconds; resize it as desired. Open the Command Line (choosing Tools... General Controls... Command Line is one way).
Choose Favorites... Add to Favorites/Toolbar to place some icons on the toolbar. This opens the Tools section of the preferences, which recapitulates Chimera's Tools menu. In the On Toolbar column, check the boxes for:
| toolbar icons | ||
|---|---|---|
|
|
|
In this tutorial, we will align the structures of three distantly related glycoside hydrolases, also used in the Images for Publication tutorial (see scientific background). If you have internet connectivity, the structures can be obtained directly from the Protein Data Bank:
Command: open 1uypIf you do not have internet connectivity, instead download the files included with this tutorial 1uyp.pdb, 1gyd.pdb, and 1oyg.pdb into your working directory and then open them in that order as local files (for example, with File... Open).
Command: open 1gyd
Command: open 1oyg
Move and scale the structures with the mouse in the graphics window and the Side View as desired throughout the tutorial.
Some salient features of the structures:
| PDB ID | enzyme | family | chains | conserved residues | ||
|---|---|---|---|---|---|---|
| 1uyp | invertase, T. maritima |
GH32 | A-F | Asp17 | Asp138 | Glu190 |
| 1gyd | arabinanase A, C. japonicus |
GH43 | B | Asp38 | Asp158 | Glu221 |
| 1oyg | levansucrase, B. subtilis |
GH68 | A | Asp86 | Asp247 | Glu342 |
Simplify the display by deleting unwanted (for our purposes) portions of the structures such as extra chains and solvent, and then displaying just the alpha-carbon traces.
Command: delete #0:.b-fSuperimposing the structures will allow examination of their commonalities and differences. The residue numbers listed above (obtained by reading the literature) could be used with the match command to specify atoms to use in a least-squares fit. However, such information is frequently not handy when one has a set of different but related structures to compare.
Command: delete solvent
Command: chain @ca
Command: linewidth 2
MatchMaker addresses this situation by first constructing a sequence alignment and then using the alpha-carbons of aligned residue pairs to match the structures. Click the icon for MatchMaker. Lacking specific knowledge of which structure might be the best Reference structure, just use the first one opened, 1uyp. The options for Chain pairing are all equivalent, because at this point, each structure has just one chain.
A variety of parameters control the sequence alignment step:
Refocus the view; show and select key conserved residues to help evaluate the fit:
Command: focus
Command: alias key #0:17.a,138.a,190.a #1:38.b,158.b,221.b #2:86.a,247.a,342.a
Command: rep sphere key
Command: select key
| structures superimposed correctly |
|---|
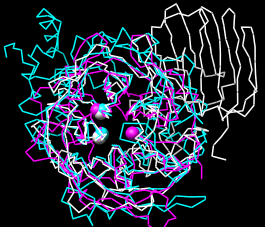 |
The selection is highlighted (by default, with green outlines) on the structures and sequence alignments. The selection highlighting makes it easy to check the sequence alignments for whether residues have been paired correctly (the correct pairings are indicated by columns in the table above). When a sequence alignment window has the mouse focus, placing the cursor over a residue in the sequence shows the associated structure residue number at the bottom of the sequence window.
If the selection is accidentally changed or erased, just select the key residues again:
Command: select keyThe selection highlighting shows that all three key residues are paired correctly in the sequence alignment of 1uyp (white) and 1gyd (magenta), while only the second two are paired correctly in the sequence alignment of 1uyp and 1oyg (cyan). However, the superposition of the structures in space is essentially correct (see the figure). MatchMaker's option to Iterate by pruning excluded incorrect parts of the sequence alignments from the fits. Match statistics are given in the Reply Log (Tools... Utilities... Reply Log). Quit from each pairwise alignment.
Use MatchMaker again, but uncheck Include secondary structure score (leaving other settings as their defaults) and click Apply. This uses a purely standard approach to sequence alignment: the Needleman-Wunsch algorithm (dynamic programming) with BLOSUM-62 scoring.
The selection highlighting now shows that none of the key residues were paired correctly in the sequence alignments. The alignment failure is understandable because the sequences are not very similar to one another. Thus, structural information in addition to sequence information is helpful for superimposing this set of structures. Quit from each pairwise alignment.
The BLOSUM-62 matrix was used to score residue similarity in the preceding calculations. Using a BLOSUM matrix intended for more distantly related structures might be expected to improve the results. However, if secondary structure scoring is turned off, BLOSUM-30 only superimposes one of the match structures (1oyg) correctly onto the reference structure, 1uyp. When combined with 30% secondary structure scoring, BLOSUM-30 and BLOSUM-62 are equally successful:
| scoring parametersa | # of pairs, RMSD (Å)b | key residue positions
aligned correctly | ||
|---|---|---|---|---|
| 1uyp vs. 1gyd | 1uyp vs. 1oyg | 1uyp vs. 1gyd | 1uyp vs. 1oyg | |
| BLOSUM-62, no secondary structure |
6, 1.276 | 7, 1.217 | 0 | 0 |
| BLOSUM-62, 30% secondary structure (MatchMaker defaults) |
71, 1.164 | 76, 1.056 | 3 | 2 |
| BLOSUM-30, no secondary structure |
49, 1.259 | 33, 0.805 | 0 | 1 |
| BLOSUM-30, 30% secondary structure |
65, 1.192 | 78, 1.059 | 3 | 2 |
Of the superpositions in this table, only those with at least one key residue position aligned correctly in the sequence alignment are correct by visual inspection.
Try various parameter settings as desired, but before proceeding with the tutorial, make sure all three structures are superimposed in a reasonably correct way (such as in the figure). Cancel the MatchMaker dialog and Quit from any existing sequence alignments.
Match -> Align can be used to construct a multiple sequence alignment from a superposition of structures. (MatchMaker produces only pairwise sequence alignments.) Click the icon for Match -> Align and Apply the default settings. Only the distances between alpha-carbons are used, not the residue types. With these settings, only sets of residues whose alpha-carbons are within 5.0 Å of each other can be placed in the same column of the output sequence alignment.
When the output appears, Close the Match -> Align dialog and check the output to see whether the key residues have been aligned correctly. The option to iterate in MatchMaker often improves superpositions by excluding any incorrect parts of the initial sequence alignments from the fit. As a result, errors in the initial pairwise alignments from MatchMaker will often be corrected in a structure-based alignment from Match -> Align. The multiple sequence alignment can be saved to a file by choosing File... Save As... from the Multalign Viewer (alignment window) menu.
Also in Multalign Viewer: if you wish, use Tools... Percent Identity to compute pairwise sequence identities (<20% for these glycoside hydrolases). Choose Structure... Select by Conservation and move the sliders to select only the residues that all three have in common (100% mavPercentConserved), then click OK. Display these residues:
Command: disp selWhen finished, end the Chimera session:
Command: stop