You're almost done already!
“Suitable for Chimera” means not too huge...
generally, depending on your computer, alignments of up to a few hundred
sequences should be fine. An alignment
(here)
of ~1000 sequences, length ~500 was OK on my desktop and laptop Macs,
but took a couple of minutes to open.
Example 1 sequence alignment:
peroxiredoxinSFLD.afa
(alignment for the
peroxiredoxin
superfamily in the Structure-Function Linkage Database, 37 sequences)
- Open both the structure and sequence alignment in Chimera. Chimera reads
several common alignment formats.
Sequences are shown in
Multalign Viewer. You can change coloring,
font size, etc. with Preferences... Appearance in that tool.
- Verify that the structure is
associated
with a sequence in the alignment. A colored rectangle will appear behind
the name of the sequence that is associated. The structure will
automatically associate with the most similar sequence if within
the mismatch tolerance (default is up to 10% of structure residues,
but this can be overruled, as shown for Example 2A).
My example alignment has a sequence that exactly matches the structure
sequence, but mismatches are fine for this purpose as long as
the register of the structure sequence with the alignment is still correct.
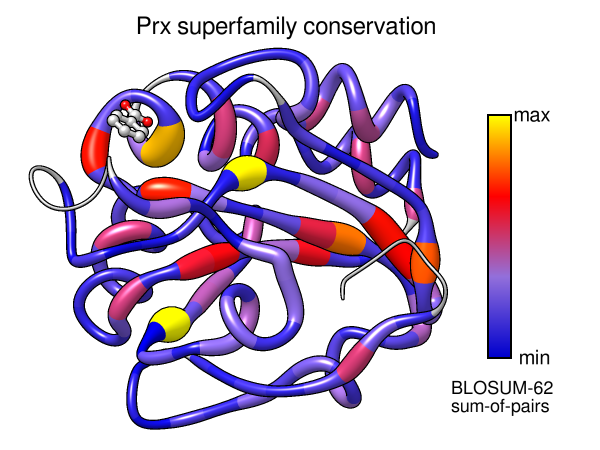
- In the Multalign Viewer window, there is a Conservation histogram
above the sequences. Choose how you want to calculate these values with the
Multalign Viewer menu: Preferences... Headers. In those preferences,
change Conservation style to AL2CO to reveal options allowing
you to choose the type of equation (entropy, variability, sum-of-pairs),
sequence weighting, and smoothing window width. As you change the settings,
the histogram above the sequences will adjust accordingly. To learn more
about AL2CO options, see:
AL2CO: calculation of positional conservation in a
protein sequence alignment.
Pei J, Grishin NV. Bioinformatics. 2001 Aug;17(8):700-12.
The SDM and HSDM matrices mentioned in this paper are only available
in Chimera 1.10 and newer.
- Again from the Multalign Viewer menu, choose: Structure...
Render by Conservation. This will call
Render by Attribute,
in which you can interactively choose colors and how they should map to
the values. The conservation values from Multalign Viewer will be the
residue attribute named mavConservation.
Try different colorings. If you go back to the previous step
and change the calculation method, then choose Refresh... Values
in the Render by Attribute menu to update its histogram
with the new values before coloring again. When you get the coloring
you like, you can turn on the option to create a color key for your figure.
You could also use “worms” (special ribbons that vary in fatness)
in addition to or instead of colors to show the conservation values
[colors+worms image].
Another way to color by attribute value is with a command, for example:
rangecolor
mavConservation -1 medium blue 0 red 3 yellow novalue white
This general process is also outlined in a
helpdesk post and the
Sequences and Structures tutorial.
You can also save the calculated conservation values to a file;
more about this below.
There are many online resources
for getting or making sequence alignments for your protein(s) of interest.
Here I'll show just a couple of examples from that long list.
Important considerations are alignment diversity
(how broad a set of sequences should be included?) and quality
(is the alignment accurate?). These may have a greater effect
on the results than the specific measure of conservation that is used.
Example 2A sequence alignment:
redoxin-seed.fasta
(seed alignment for the
redoxin family
in PFAM, 68 sequences;
the full alignment of nearly 10K sequences is too big for Chimera)
How did I know this PFAM family goes with structure 1HD2?
One way is to look at the RCSB PDB entry
1HD2 “External Domain Annotations.”
Another way is to search PFAM for “1hd2”
and then look at the “Sequence mapping” for that
structure entry.
I prefer to get the FASTA format from PFAM, as the additional
annotations in Stockholm format make the file bigger and sometimes
cause problems. PFAM alignments may include blank columns,
which can be removed in Chimera or
Jalview.
Issue: none of the sequences in the alignment are similar enough to the
structure sequence to associate automatically. Possible solutions:
- Add the sequence of the structure to the alignment. Multalign Viewer menu:
Edit... Add Sequence, From Structure tab.
If the structure sequence is difficult to align with the others,
this may require dorking around with parameters and tedious cycles
of adding and removing the sequence (Edit... Delete Sequences/Gaps)
to get it aligned properly. (There is also Edit... Realign Sequences
to realign everything, but you might not want to alter your input alignment.)
- Force association to the best-matching sequence. Multalign Viewer menu:
Structure... Associations. In this example, even though the best match
(sequence Q8MUN0_PYRRU) has 74 mismatches, the remaining >50% sequence ID
is enough to get the correct register of the structure with the alignment.
- Find a structure more similar to at least one of the sequences in
the alignment (not always easy). For example, PDB
1XIY
associates automatically with sequence Q5MYR6_PLAF7 with just 2 mismatches.
The list of structures for the redoxin family at PFAM shows that this
structure and sequence go together.
Then you can proceed as in Case 1.
Example 2B Chimera session:
ConSurf-1hd2-chimera19.py
[show this last!]
(result of submitting 1hd2 to the ConSurf server and choosing to show results in Chimera;
alignment has 150 sequences including that of the query, 1hd2)
Given a protein structure, the
ConSurf server
estimates the evolutionary conservation of amino acid positions based on
the phylogenetic relations between homologous sequences
(details...). It also works on nucleic acids.
Choosing to show the results in Chimera will download a *.chimerax
(Chimera web data)
file, which in turn references URLs for several files of results at the
ConSurf website. Opening the chimerax file loads everything into Chimera:
the structure and a sequence alignment both colored by ConSurf conservation
scores. The alignment includes a phylogenetic tree representation on the left
and ConSurf scores as two custom alignment headers (integers and histogram)
across the top, as shown in the figure.
Best to save a Chimera session with these results, since they
won't be kept forever at the ConSurf website.
Besides using the ConSurf scores, you can still show the Chimera
Conservation header for the ConSurf alignment
(turn it on using the Headers menu in Multalign Viewer),
apply any of the AL2CO methods, and render attribute
mavConservation, as in Case 1.
You can un-show the tree using the Tree menu, and of course
change the display style of the structure.
See another
ConSurf example with more details on the chimerax and results files.
There are two general ways to assign arbitrary per-residue values in Chimera.
Both require putting the values into a relatively simple text file format
that can be read into Chimera (example files below):
- Assign values directly to structure residues via an
attribute assignment file
Pros: you don't need to have a sequence alignment for Chimera;
the process is general for assigning any set(s) of values to atoms or residues
for easy visualization
(general examples)
Cons: you have to specify the target atoms or residues,
and if using residue numbers, different assignment files would be needed
for structures with different numbering (and for alignment-derived values,
different placement of gaps/insertions relative to that alignment).
If multiple structures are open, one should be careful to assign the values
to the intended structure only.
- Create a custom header
for your sequence alignment; numeric headers are automatically propagated
as a residue attribute of any structure(s) associated with the alignment
Pros: you can display the values as a histogram over the alignment,
and they will be assigned automatically as residue attributes
of any associated structures, regardless of how they are aligned
or numbered (association takes care of the sequence-structure mapping)
Cons: requires a corresponding sequence alignment that Chimera can show
Example 4A residue attribute assignment files:
How to use these files:
- open the corresponding structure in Chimera
- read in the attribute file using
Define Attribute
(in menu under Tools... Structure Analysis) or the command
defattr,
being careful to apply the values to the correct structure
- then your new custom attributes will appear in
Render by Attribute
for coloring, etc. as shown in the figure.
How I made the files...
alignment with Conservation (entropy measure),
other headers loaded from the two example files |
 |
Example 4B alignment header files
(for redoxin-seed.fasta):
- redoxin-seed-HSDM-norm.txt
- conservation calculated with the HSDM matrix, with some coloring
just as an example
- redoxin-anno.txt
- a whole slew of conservation measures calculated in
Jalview using AACon,
processed and reformatted into the Chimera header format. You can see
from the figure that most of these measures suffer from poor handling of
high-gap-fraction columns.
How to use these files:
- open the corresponding sequence alignment, in this case
redoxin-seed.fasta
- use Multalign Viewer menu: Headers... Load to open the file(s)
- if any structures are associated with the alignment, residue attributes
corresponding to your custom numerical headers will be available in
Render by Attribute
for coloring, etc. as shown in the figure.
How I made the files...
Custom headers can also include symbols
[image].






{kind=link}
{kind=link}