Tom Goddard
January 10, 2025
Macromethods course team project
Here are steps to setup predictions with AlphaFold 3 of FDA prescription drug compounds with a target viral protein to run on the UCSF Wynton compute cluster.
We have 3 teams in the macromethods course and I suggest each work on a different virus: Avian Influenza, Monkey Pox, Respiratory syncytial virus (RSV). Each team member can choose their own virus protein. It is ok for more than one team member to choose the same protein -- they will get different predictions due to the random seed used in the predictions. Teams decide which virus they want to work with.
Because we have limited time for this project choose a protein under 350 amino acids. The AlphaFold 3 runtime increases rapidly with protein size (as the square of the number of amino acids). The proteins for each virus are listed on these UniProt sequence database web pages.
Download the sequence for the protein you choose from the UniProt entry page for the gene to your laptop. The download link is near the bottom of the page in the UniProt page "Sequence" section and saves as a .fasta text file.
Create an AlphaFold 3 input text file in json format with a name that indicates your virus and protein, for instance here is an mpox_d10.json example:
{
"name": "mpox_d10",
"modelSeeds": [1],
"sequences": [
{"protein":
{"id": "A",
"sequence": "MSFYRSSIISQIIKYNRRLAKSIICEDDSQIITLTAFVNQCLWCHKRVSVSAILLTTDNKILVCNRRDSFLYSEIIRTRNMYRKKRLFLNYSNYLNKQERSILSSFFSLDPATADNDRINAIYPGGIPKRGENVPECLSREIKEEVNIDNSFVFIDTRFFIHGIIEDTIINKFFEVIFFVGRISLTSDQIIDTFKSNHEIKDLIFLDPNSGNGLQYEIAKYALDTAKLKCYGHRGCYYESLKKLTEDD"
}
}
],
"dialect": "alphafold3",
"version": 1
}
You can use this as a template. Change the "name" value -- that is the name of the directory that AlphaFold 3 will create for the prediction results. Change the "modelSeeds" value from 1 to some positive integer. This is used to generate random numbers and different seed values will give different results. Change the "sequence" value to the amino acid sequence all on one line in quotes.
The command to submit the AlphaFold prediction job on Wynton looks like this
qsub -l h_rt=2:00:00 /wynton/home/ferrin/goddard/alphafold_singularity/run_alphafold3.py --json_path=mpox_d10.json
I put this command in a text file named "command" so that later I will be able to find exactly what command I used for this prediction.
You need the AlphaFold 3 "weights" which are the neural network parameters obtained from Google by filling out a form linked on the AlphaFold 3 github page in the Obtaining Model Parameters section. They say that takes 2-3 days. If you have the parameters af3.bin.zst copy them to your Wynton home directory and put them in a directory ~/af3_weights, my run_alphafold3.py script uses that default location for the parameters.
From your laptop where you downloaded the parameters from Google:
$ rsync -av af3.bin.zst goddard@log1.wynton.ucsf.edu:
Then on Wynton make the ~/af3_weights directory and put the uncompressed parameters af3.bin in that directory
$ ssh goddard@log1.wynton.ucsf.edu
$ mkdir af3_weights
$ mv af3.bin.zst af3_weights
$ cd af3_weights
$ zstd -d af3.bin.zst
$ rm af3.bin.zst
If you have not received the AlphaFold 3 model parameters from Google you can use mine. Remember they are for non-commercial use only. Copy them from my Wynton directory to yours
$ cd
$ rsync -av ~goddard/af3_weights .
Put your .fasta, .json and command files in a directory, for this example I named the directory mpox_d10. Then copy them to your Wynton home directory using shell command (for instance use Mac /Applications/Utilities/Terminal):
$ cd
$ rsync -av mpox_d10 goddard@log1.wynton.ucsf.edu:
Login to your Wynton account. Change to the directory that you copied for the prediction then paste the qsub command to the shell prompt.
$ ssh goddard@log1.wynton.ucsf.edu
$ cd mpox_d10
$ qsub -l h_rt=2:00:00 /wynton/home/ferrin/goddard/alphafold_singularity/run_alphafold3.py --json_path=mpox_d10.json
Use the SGE command qstat to see if your AF3 prediction job has started
$ qstat
job-ID prior name user state submit/start at queue slots ja-task-ID
-----------------------------------------------------------------------------------------------------------------
1765394 0.27120 alphafold goddard r 01/10/2025 17:33:40 gpu.q@qb3-atgpu21 1
If there is a "qw" in the "state" column that means it is in queue wait, waiting to run. If there is an "r" that means it has started running. In my test on January 10 at about 5 pm it took about 6 minutes before the job started running. This will depend on how many other jobs are waiting to run. It could take an hour to start.
If qstat returns nothing then the job is done. That may mean it finished successfully or failed. See the next section for details.
When the jobs runs it will produce standard output and standard error text files named like
alphafold.e1765394
alphafold.o1765394
The number at the end is the job number. Looking at the alphafold.e1765394 file will show the progress. Here is an example where it is part way through the initial sequence alignment step after about 50 minutes.
$ cat alphafold.e1765394
mkdir: cannot create directory '/tmp/lock-gpu1': File exists
mkdir: cannot create directory '/tmp/lock-gpu2': File exists
mkdir: cannot create directory '/tmp/lock-gpu3': File exists
I0110 17:33:50.681979 23047588233216 xla_bridge.py:895] Unable to initialize backend 'rocm': module 'jaxlib.xla_ex\
tension' has no attribute 'GpuAllocatorConfig'
I0110 17:33:50.703902 23047588233216 xla_bridge.py:895] Unable to initialize backend 'tpu': INTERNAL: Failed to op\
en libtpu.so: libtpu.so: cannot open shared object file: No such file or directory
I0110 17:33:50.800672 23047588233216 folding_input.py:1115] Detected mpox_d10.json is an AlphaFold 3 JSON since th\
e top-level is not a list.
2025-01-10 17:33:51.519787: W external/xla/xla/service/gpu/nvptx_compiler.cc:930] The NVIDIA driver's CUDA version\
is 12.4 which is older than the PTX compiler version 12.6.77. Because the driver is older than the PTX compiler v\
ersion, XLA is disabling parallel compilation, which may slow down compilation. You should update your NVIDIA driv\
er or use the NVIDIA-provided CUDA forward compatibility packages.
I0110 17:33:54.896365 23047588233216 pipeline.py:81] Getting protein MSAs for sequence MSFYRSSIISQIIKYNRRLAKSIICED\
DSQIITLTAFVNQCLWCHKRVSVSAILLTTDNKILVCNRRDSFLYSEIIRTRNMYRKKRLFLNYSNYLNKQERSILSSFFSLDPATADNDRINAIYPGGIPKRGENVPECLSRE\
IKEEVNIDNSFVFIDTRFFIHGIIEDTIINKFFEVIFFVGRISLTSDQIIDTFKSNHEIKDLIFLDPNSGNGLQYEIAKYALDTAKLKCYGHRGCYYESLKKLTEDD
I0110 17:33:54.900367 23036604438080 jackhmmer.py:78] Query sequence: MSFYRSSIISQIIKYNRRLAKSIICEDDSQIITLTAFVNQCLWC\
HKRVSVSAILLTTDNKILVCNRRDSFLYSEIIRTRNMYRKKRLFLNYSNYLNKQERSILSSFFSLDPATADNDRINAIYPGGIPKRGENVPECLSREIKEEVNIDNSFVFIDTR\
FFIHGIIEDTIINKFFEVIFFVGRISLTSDQIIDTFKSNHEIKDLIFLDPNSGNGLQYEIAKYALDTAKLKCYGHRGCYYESLKKLTEDD
I0110 17:33:54.900514 23036602336832 jackhmmer.py:78] Query sequence: MSFYRSSIISQIIKYNRRLAKSIICEDDSQIITLTAFVNQCLWC\
HKRVSVSAILLTTDNKILVCNRRDSFLYSEIIRTRNMYRKKRLFLNYSNYLNKQERSILSSFFSLDPATADNDRINAIYPGGIPKRGENVPECLSREIKEEVNIDNSFVFIDTR\
FFIHGIIEDTIINKFFEVIFFVGRISLTSDQIIDTFKSNHEIKDLIFLDPNSGNGLQYEIAKYALDTAKLKCYGHRGCYYESLKKLTEDD
I0110 17:33:54.900583 23036606539328 jackhmmer.py:78] Query sequence: MSFYRSSIISQIIKYNRRLAKSIICEDDSQIITLTAFVNQCLWC\
HKRVSVSAILLTTDNKILVCNRRDSFLYSEIIRTRNMYRKKRLFLNYSNYLNKQERSILSSFFSLDPATADNDRINAIYPGGIPKRGENVPECLSREIKEEVNIDNSFVFIDTR\
FFIHGIIEDTIINKFFEVIFFVGRISLTSDQIIDTFKSNHEIKDLIFLDPNSGNGLQYEIAKYALDTAKLKCYGHRGCYYESLKKLTEDD
I0110 17:33:54.900846 23036600235584 jackhmmer.py:78] Query sequence: MSFYRSSIISQIIKYNRRLAKSIICEDDSQIITLTAFVNQCLWC\
HKRVSVSAILLTTDNKILVCNRRDSFLYSEIIRTRNMYRKKRLFLNYSNYLNKQERSILSSFFSLDPATADNDRINAIYPGGIPKRGENVPECLSREIKEEVNIDNSFVFIDTR\
FFIHGIIEDTIINKFFEVIFFVGRISLTSDQIIDTFKSNHEIKDLIFLDPNSGNGLQYEIAKYALDTAKLKCYGHRGCYYESLKKLTEDD
I0110 17:33:54.901517 23036604438080 subprocess_utils.py:68] Launching subprocess "/hmmer/bin/jackhmmer -o /dev/nu\
ll -A /scratch/1765394.1.gpu.q/tmp4vovfhxt/output.sto --noali --F1 0.0005 --F2 5e-05 --F3 5e-07 --cpu 8 -N 1 -E 0.\
0001 --incE 0.0001 /scratch/1765394.1.gpu.q/tmp4vovfhxt/query.fasta /wynton/group/databases/alphafold3/mgy_cluster\
s_2022_05.fa"
I0110 17:33:54.902037 23036602336832 subprocess_utils.py:68] Launching subprocess "/hmmer/bin/jackhmmer -o /dev/nu\
ll -A /scratch/1765394.1.gpu.q/tmpl9ldkim_/output.sto --noali --F1 0.0005 --F2 5e-05 --F3 5e-07 --cpu 8 -N 1 -E 0.\
0001 --incE 0.0001 /scratch/1765394.1.gpu.q/tmpl9ldkim_/query.fasta /wynton/group/databases/alphafold3/bfd-first_n\
on_consensus_sequences.fasta"
I0110 17:33:54.902811 23036606539328 subprocess_utils.py:68] Launching subprocess "/hmmer/bin/jackhmmer -o /dev/nu\
ll -A /scratch/1765394.1.gpu.q/tmpd9a12wso/output.sto --noali --F1 0.0005 --F2 5e-05 --F3 5e-07 --cpu 8 -N 1 -E 0.\
0001 --incE 0.0001 /scratch/1765394.1.gpu.q/tmpd9a12wso/query.fasta /wynton/group/databases/alphafold3/uniref90_20\
22_05.fa"
I0110 17:33:54.903393 23036600235584 subprocess_utils.py:68] Launching subprocess "/hmmer/bin/jackhmmer -o /dev/nu\
ll -A /scratch/1765394.1.gpu.q/tmpl81hfsni/output.sto --noali --F1 0.0005 --F2 5e-05 --F3 5e-07 --cpu 8 -N 1 -E 0.\
0001 --incE 0.0001 /scratch/1765394.1.gpu.q/tmpl81hfsni/query.fasta /wynton/group/databases/alphafold3/uniprot_all\
_2021_04.fa"
I0110 17:36:07.394335 23036602336832 subprocess_utils.py:97] Finished Jackhmmer in 132.490 seconds
I0110 17:41:42.297356 23036606539328 subprocess_utils.py:97] Finished Jackhmmer in 467.392 seconds
I0110 17:49:55.463403 23036604438080 subprocess_utils.py:97] Finished Jackhmmer in 960.560 seconds
When the job finishes "qstat" returns nothing. In this example it took 59 minutes. Hopefully there are no errors at the end of the standard error file alphafold.e1765394 (there may be some warnings about (invalid value encoutnred in divide) which can probably be ignored) and the standard output file alphafold.o1765394 looks something like this
$ cat alphafold.o1765394
Running AlphaFold 3. Please note that standard AlphaFold 3 model parameters are
only available under terms of use provided at
https://github.com/google-deepmind/alphafold3/blob/main/WEIGHTS_TERMS_OF_USE.md.
If you do not agree to these terms and are using AlphaFold 3 derived model
parameters, cancel execution of AlphaFold 3 inference with CTRL-C, and do not
use the model parameters.
Found local devices: [CudaDevice(id=0)]
Building model from scratch...
Processing fold inputs.
Processing fold input #1
Processing fold input mpox_d10
Checking we can load the model parameters...
Running data pipeline...
Processing chain A
Processing chain A took 3089.39 seconds
Output directory: ./mpox_d10
Writing model input JSON to ./mpox_d10
Predicting 3D structure for mpox_d10 for seed(s) (1,)...
Featurising data for seeds (1,)...
Featurising mpox_d10 with rng_seed 1.
Featurising mpox_d10 with rng_seed 1 took 3.53 seconds.
Featurising data for seeds (1,) took 8.51 seconds.
Running model inference for seed 1...
Running model inference for seed 1 took 45.41 seconds.
Extracting output structures (one per sample) for seed 1...
Extracting output structures (one per sample) for seed 1 took 0.22 seconds.
Running model inference and extracting output structures for seed 1 took 45.63 seconds.
Running model inference and extracting output structures for seeds (1,) took 45.63 seconds.
Writing outputs for mpox_d10 for seed(s) (1,)...
Done processing fold input mpox_d10.
Done processing 1 fold inputs.
singularity exec --nv -B "/wynton/group/databases/alphafold3,/wynton/home/ferrin/goddard/af3_weights,/wynton/home/\
ferrin/goddard/mpox_d10,/scratch/1765394.1.gpu.q" --env CUDA_VISIBLE_DEVICES=0,NVIDIA_VISIBLE_DEVICES=0 /wynton/ho\
me/ferrin/goddard/alphafold_singularity/alphafold3_40gb_dec_4_2024.sif python /app/alphafold/run_alphafold.py --js\
on_path=mpox_d10.json --db_dir=/wynton/group/databases/alphafold3 --output_dir=. --model_dir=/wynton/home/ferrin/g\
oddard/af3_weights --flash_attention_implementation=triton --run_data_pipeline=True --run_inference=True --num_dif\
fusion_samples=5 --jackhmmer_n_cpu=8 --nhmmer_n_cpu=8
Copy the prediction results back from Wynton to your laptop. On your laptop run
$ cd
$ rsync -av goddard@log1.wynton.ucsf.edu:mpox_d10 .
mpox_d10/alphafold.o1765394
mpox_d10/mpox_d10.json
mpox_d10/mpox_d10/
mpox_d10/mpox_d10/TERMS_OF_USE.md
mpox_d10/mpox_d10/mpox_d10_confidences.json
mpox_d10/mpox_d10/mpox_d10_data.json
mpox_d10/mpox_d10/mpox_d10_model.cif
mpox_d10/mpox_d10/mpox_d10_summary_confidences.json
mpox_d10/mpox_d10/ranking_scores.csv
mpox_d10/mpox_d10/seed-1_sample-0/
mpox_d10/mpox_d10/seed-1_sample-0/confidences.json
mpox_d10/mpox_d10/seed-1_sample-0/model.cif
mpox_d10/mpox_d10/seed-1_sample-0/summary_confidences.json
mpox_d10/mpox_d10/seed-1_sample-1/
mpox_d10/mpox_d10/seed-1_sample-1/confidences.json
mpox_d10/mpox_d10/seed-1_sample-1/model.cif
mpox_d10/mpox_d10/seed-1_sample-1/summary_confidences.json
mpox_d10/mpox_d10/seed-1_sample-2/
mpox_d10/mpox_d10/seed-1_sample-2/confidences.json
mpox_d10/mpox_d10/seed-1_sample-2/model.cif
mpox_d10/mpox_d10/seed-1_sample-2/summary_confidences.json
mpox_d10/mpox_d10/seed-1_sample-3/
mpox_d10/mpox_d10/seed-1_sample-3/confidences.json
mpox_d10/mpox_d10/seed-1_sample-3/model.cif
mpox_d10/mpox_d10/seed-1_sample-3/summary_confidences.json
mpox_d10/mpox_d10/seed-1_sample-4/
mpox_d10/mpox_d10/seed-1_sample-4/confidences.json
mpox_d10/mpox_d10/seed-1_sample-4/model.cif
mpox_d10/mpox_d10/seed-1_sample-4/summary_confidences.json
|
|
|
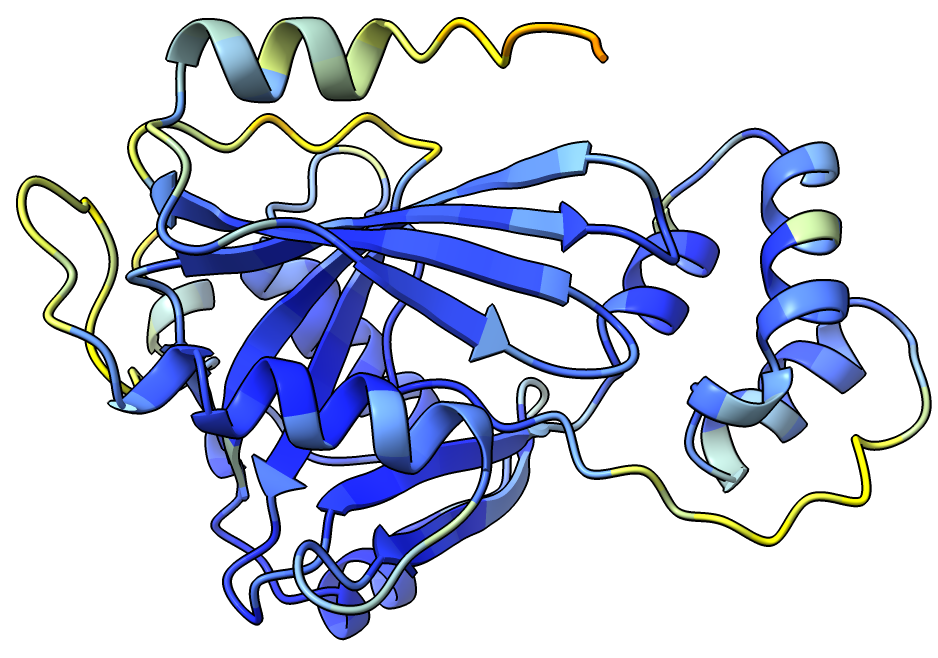
Start ChimeraX on your laptop and open the predicted atomic model mpox_d10/mpox_d10/mbox_d10_model.cif. You can color using the AlphaFold 3 predicted per-residue confidence (pLDDT) blue high confidence, yellow-red low confidence with ChimeraX command (AlphaFold writes the pLDDT scores as the bfactor)
color bfactor palette alphafold
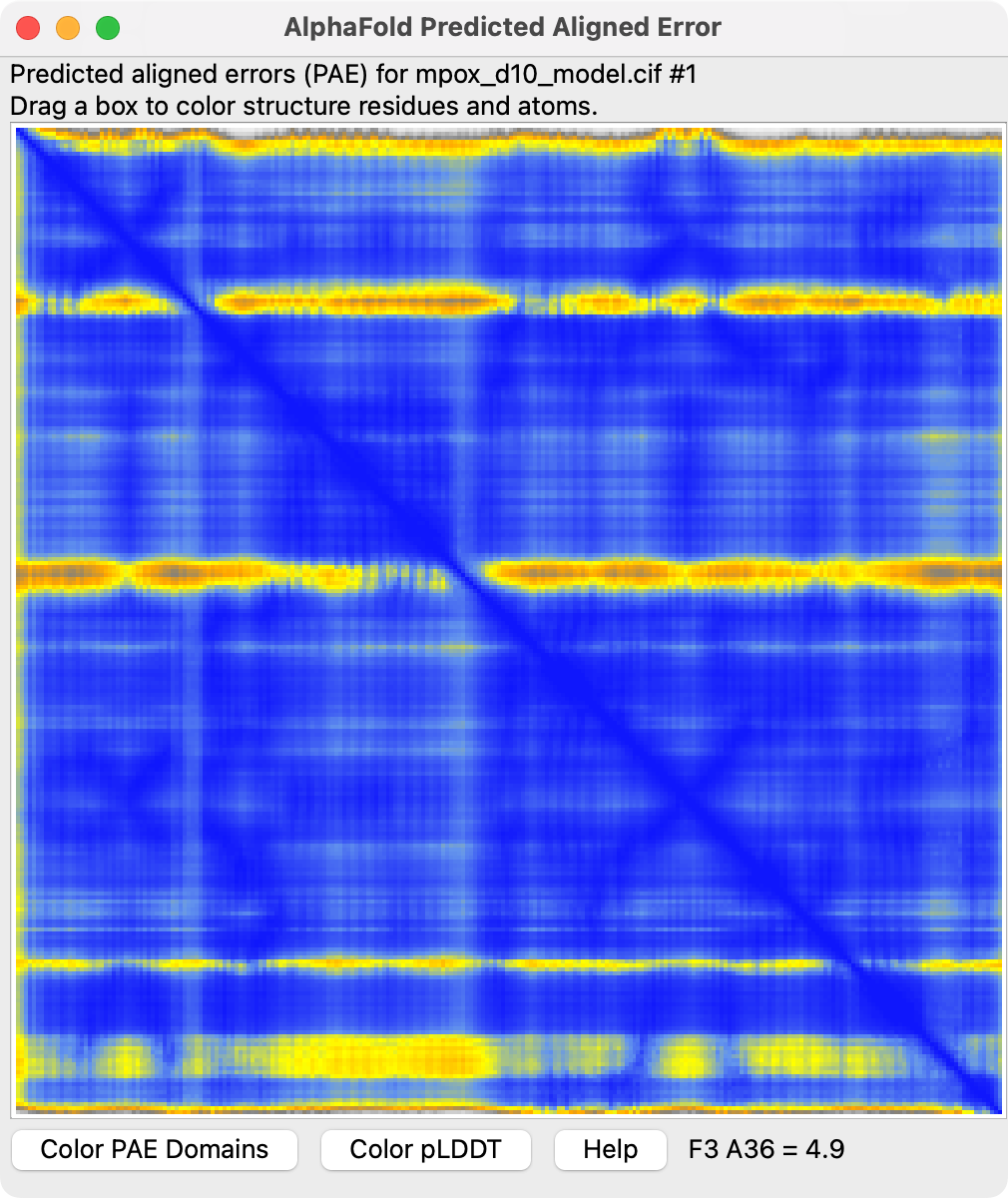
You can show the predicted aligned error confidence score which is a score between every pair of residues saying whether the relative alignment of the two residues is good using ChimeraX menu Tools / Structure Prediction / AlphaFold Error Plot.

| 
| 
|
While waiting an hour for the prediction to complete we can look at some example prediction results using Nipah virus G protein with two zanamivir ligands wynton_nipah_zmr.zip.
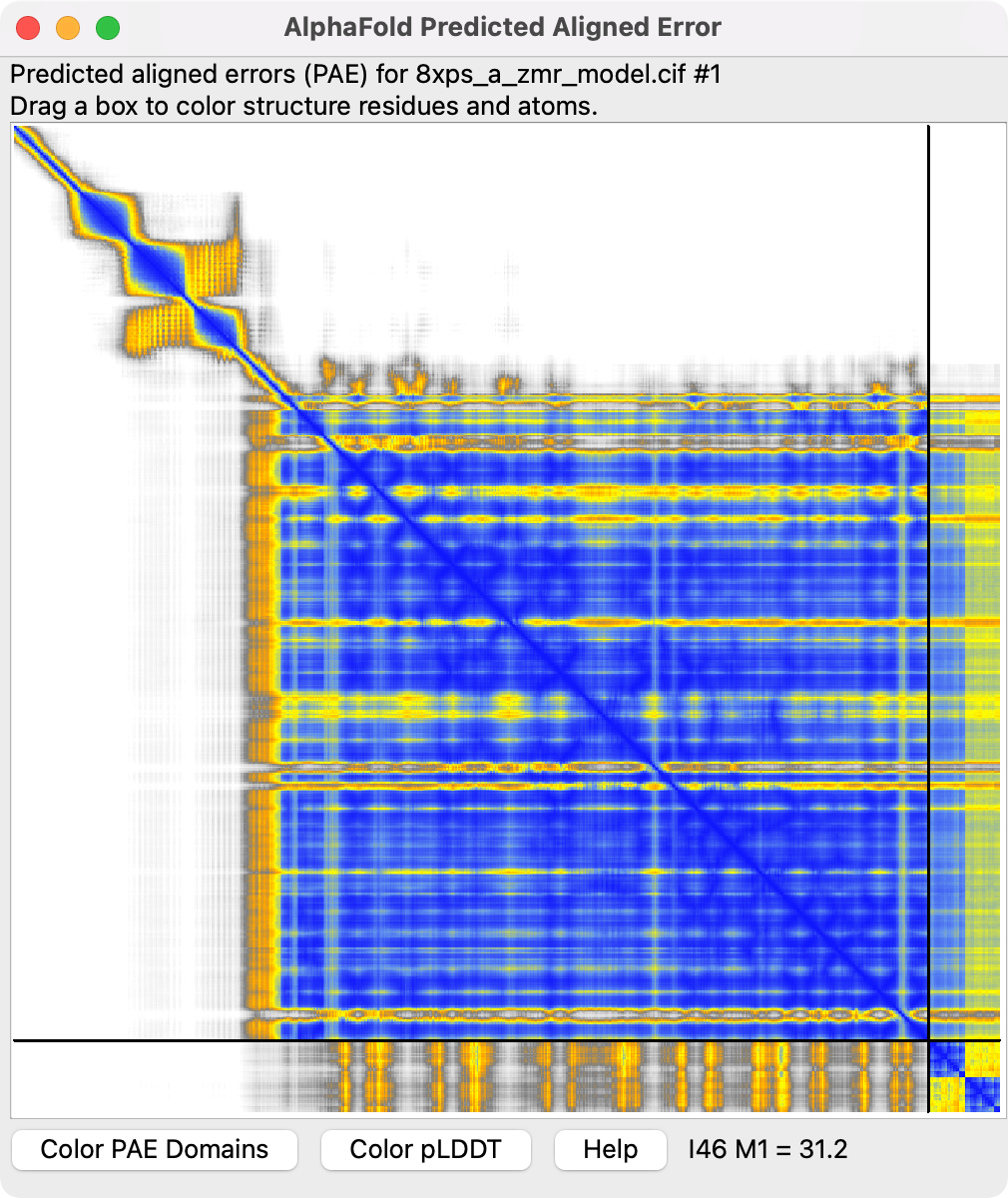
I predicted with two copies of zanamivir because homolog influenza neuraminidase structure PDB 2CML has two zanamivirs bound at different sites. In ChimeraX we can compare this structure to the Nipah prediction.
open 2cml
delete #2 & ~/A
hide #2 & ~:ZMR atoms,ribbon
color #2:ZMR & C magenta
We see the two predicted zanamivirs are stacked near one of the influenza zanamivirs colored magenta. We also see the AlphaFold 3 assigns low confidence to the predicted ligands since they are colored yellow, meaning they have a low predicted local distance difference test (pLDDT) score. We can Also in the PAE plot the lower right corner shows the two ligands (blue squares) but the columns and rows for those are yellow indicating poor predicted aligned error (PAE) scrores.
| Chain pair ipTM scores | ||
| 0.71 | 0.85 | 0.74 |
| 0.85 | 0.27 | 0.2 |
| 0.74 | 0.2 | 0.28 |
Our strategy is to make a separate AlphaFold 3 input file for each drug that lists both the drug (as a SMILES string) and virus protein, then ask AlphaFold 3 to predict the structures for every input file in the directory.
We will use this list fda_smiles.txt of 1509 active ingredients of FDA approved prescription drugs. The file gives the compound name and the SMILES string which describes the chemical structure.
Make a directory for the input files
$ cd
$ mkdir mpox_d10_drugs
Copy the virus protein input file to this new directory. When we ran AlphaFold 3 with just the virus protein it produced a new JSON input file that includes a deep multiple sequence alignment (MSA) and also template structures found in the PDB. We will use that new input file so AlphaFold does not have to recompute those which takes 95% of the computation time.
$ cp mpox_d10/mpox_d10/mpox_d10_data.json mpox_d10_drugs/mpox_d10_with_msa.json
To avoid copying the MSA and templates for each of the 1509 drug input files we will separate them from the mpox_d10_with_msa.json file using a Python script I wrote af3_extract_msa.py. This creates additional files for the MSA and templates and changes the .json file to point to those additional files.
$ cd mpox_d10_drugs
$ python3 af3_extract_msa.py mpox_d10_with_msa.json mpox_d10_apo.json
$ ls
mpox_d10_apo.json
mpox_d10_apo_paired_msa_A.a3m
mpox_d10_apo_template_A_1.cif
mpox_d10_apo_template_A_2.cif
mpox_d10_apo_template_A_3.cif
mpox_d10_apo_template_A_4.cif
mpox_d10_apo_unpaired_msa_A.a3m
mpox_d10_with_msa.json
Now use another Python script af3_drug_json_input.py that makes a new input file for each drug by adding the drug to mpox_d10_apo.json. The new input files will be named using the drug name.
$ python3 af3_drug_json_input.py mpox_d10_apo.json fda_smiles.txt
$ ls
abacavir_sulfate.json
abametapir.json
abemaciclib.json
abiraterone_acetate.json
abrocitinib.json
...
The command to run all the predictions has a few different options. I put this command text in a file named "command" so that later I can find exactly the command I ran.
qsub -l h_rt=36:00:00 /wynton/home/ferrin/goddard/alphafold_singularity/run_alphafold3.py --input_dir . --run_data_pipeline false --num_diffusion_samples 2
Copy the input files from your laptop to Wynton
$ cd
$ rsync -a mpox_d10_drugs goddard@log1.wynton.ucsf.edu:
$ ssh goddard@log1.wynton.ucsf.edu
$ cd mpox_d10_drugs
$ qsub -l h_rt=36:00:00 /wynton/home/ferrin/goddard/alphafold_singularity/run_alphafold3.py --input_dir . --run_data_pipeline false --num_diffusion_samples 2
Use the qstat command as described above to see when the job starts as described above. It is good to keep an eye on it until it starts because it might fail as soon as it starts if you typed something wrong, and you'll want to fix it and restart.
Check the progress by looking in the alphafold.e1234567 file after the job is running. For this example the predictions took 15 hours on an A40 GPU.
When the prediction finishes and is successful copy the prediction results back from Wynton to your laptop. This uses 4.5 Gbytes of disk space and takes several minutes to copy the files. On your laptop run
$ cd
$ rsync -av goddard@log1.wynton.ucsf.edu:mpox_d10_drugs .
I wrote a ChimeraX Python script drug_scores.py to go through the 1509 predictions and extract confidence scores. It reads the interface predicted TM score (ipTM), the average pLDDT score for ligand atoms, and looks at the number of high confidence PAE scores between ligand atoms and nearby residues. Download the drug_scores.py script and put it in your predictions directory mpox_d10_drugs. Then start ChimeraX and run the script on the predicted structures from the ChimeraX command-line
cd ~/mpox_d10_drugs
runscript drug_scores.py */*_model.cif
This will take five or more minutes to load each of the 1509 predictions and accumulate the scores and print them to the ChimeraX log panel sorted by highest scores at the top. Copy and paste these scores to a text file mpox_d10_scores.txt.
Name ipTM pLDDT PAE<3A/dist<4A/natom,nres,npair estradiol 0.96 95 14 12 29 iptacopan_hydrochloride 0.95 95 28 18 46 efavirenz 0.95 93 20 15 48 doxycycline 0.95 94 32 14 66 citalopram_hydrobromide 0.95 89 19 13 34 chlordiazepoxide 0.95 94 16 14 40 bumetanide 0.95 91 23 13 48 metformin_hydrochloride 0.94 88 8 6 18 ...
Here is some info about the top scoring hits:
Four out of the top 5 bind in approximately the same location, with citalopram_hydrobromide binding at a different site. The chlorine in efavirenz has become detached in the prediction.
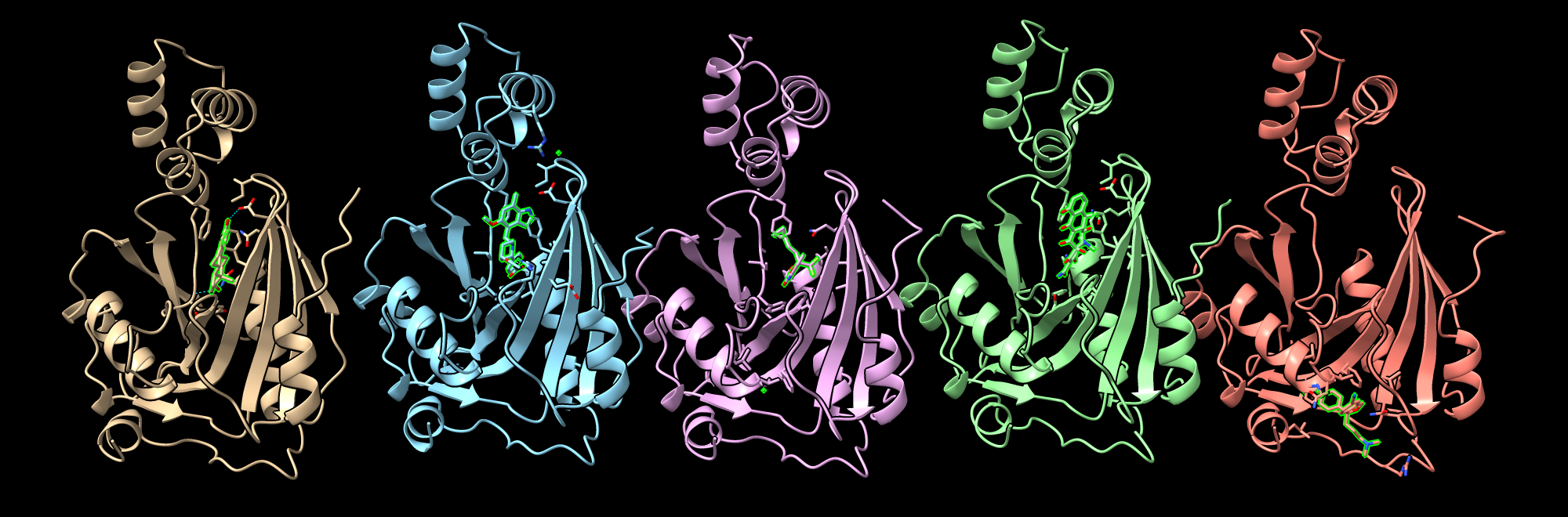
| 
| 
|
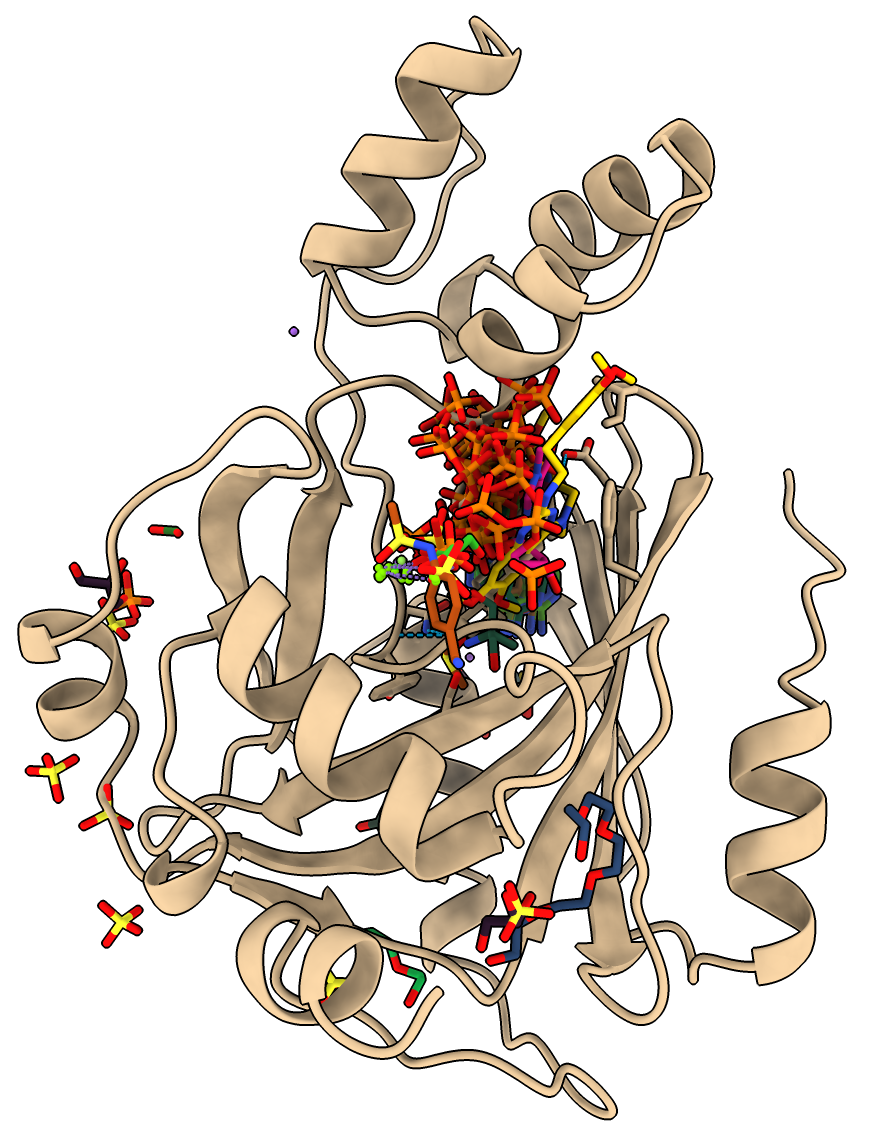
Using the ChimeraX Foldseek search (menu Tools / Structure Analysis / Similar Structures) and the Ligands button on that tool finds some related structures and using the top hit estradiol mpox_d10 as the query (tan) finds 3 similar PDB models with bound ligands:

Would be nice to know if any of the predicted FDA compounds are similar to these ligands found in the PDB.
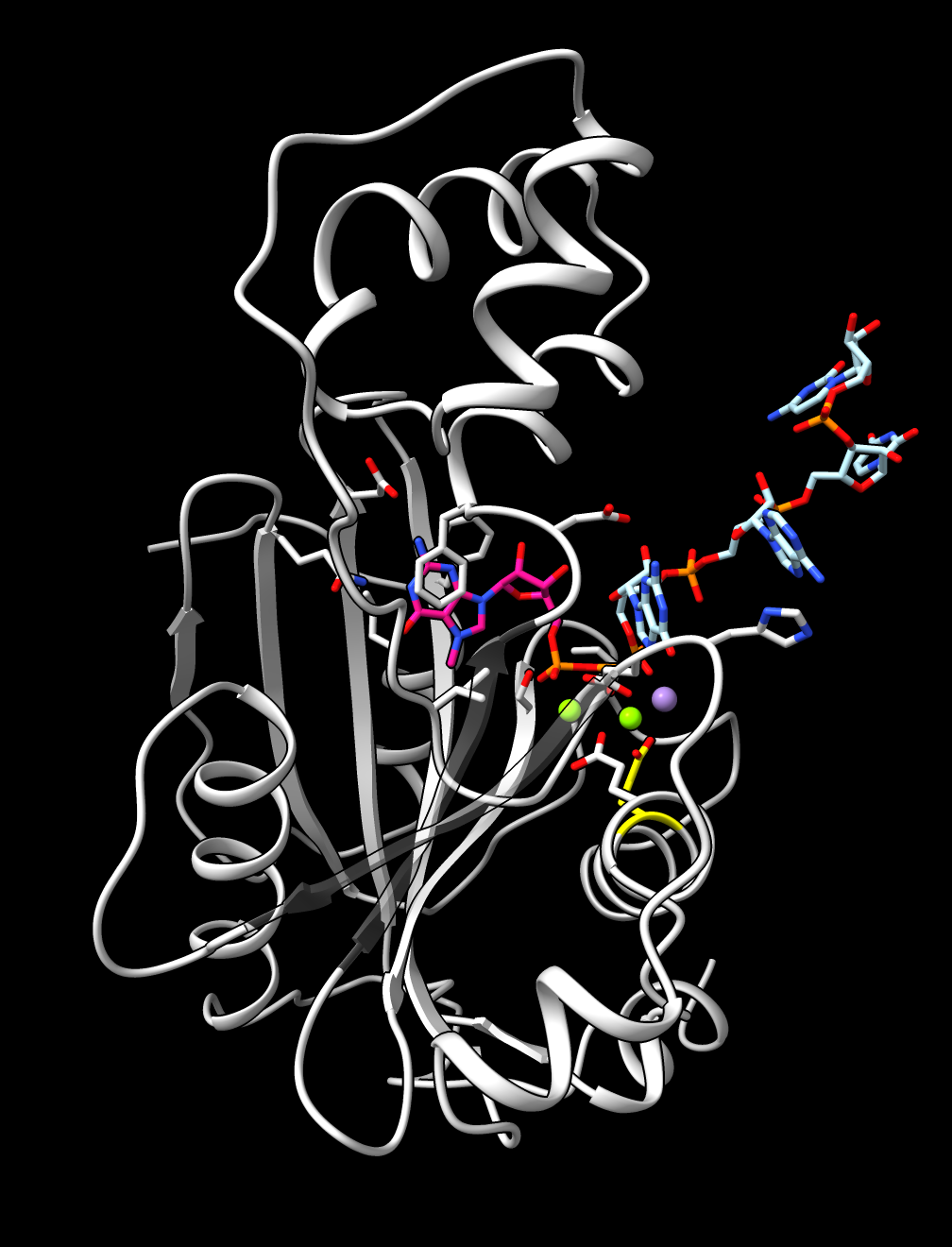
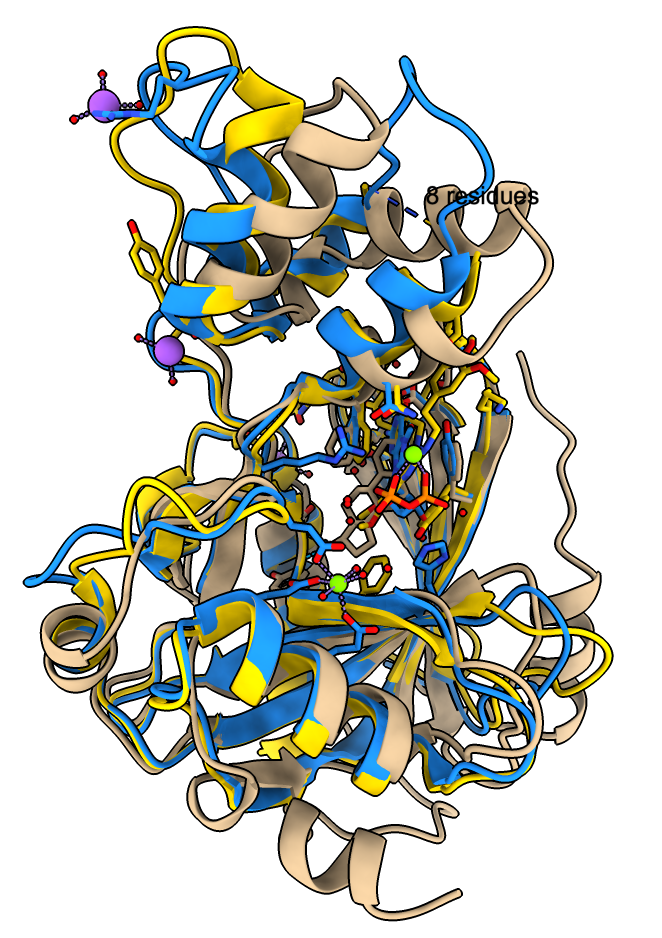
AF3 prediction of monkeypox OPG122 (E10R) with a 5' capped mRNA. | 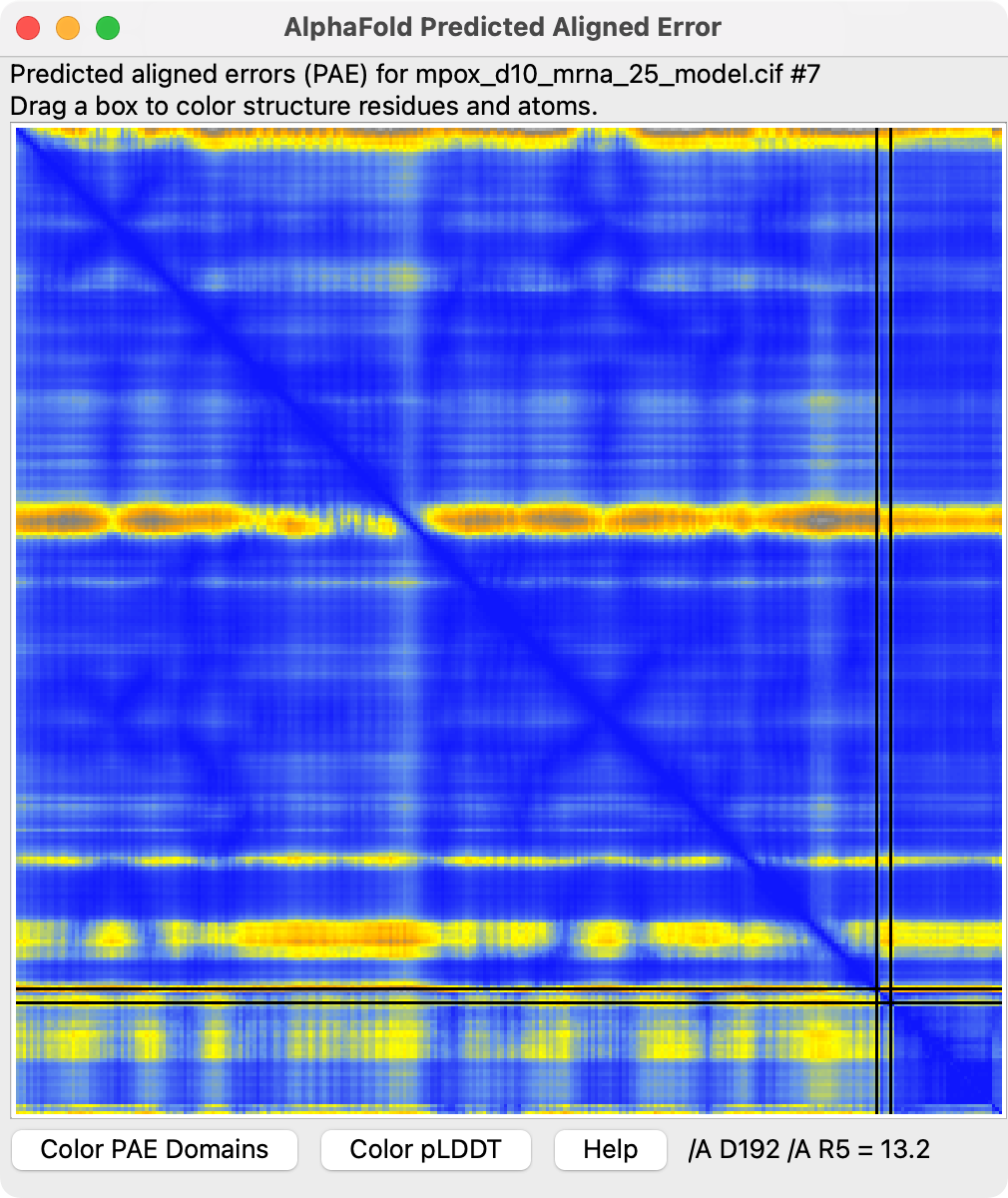
Predicted aligned error. | 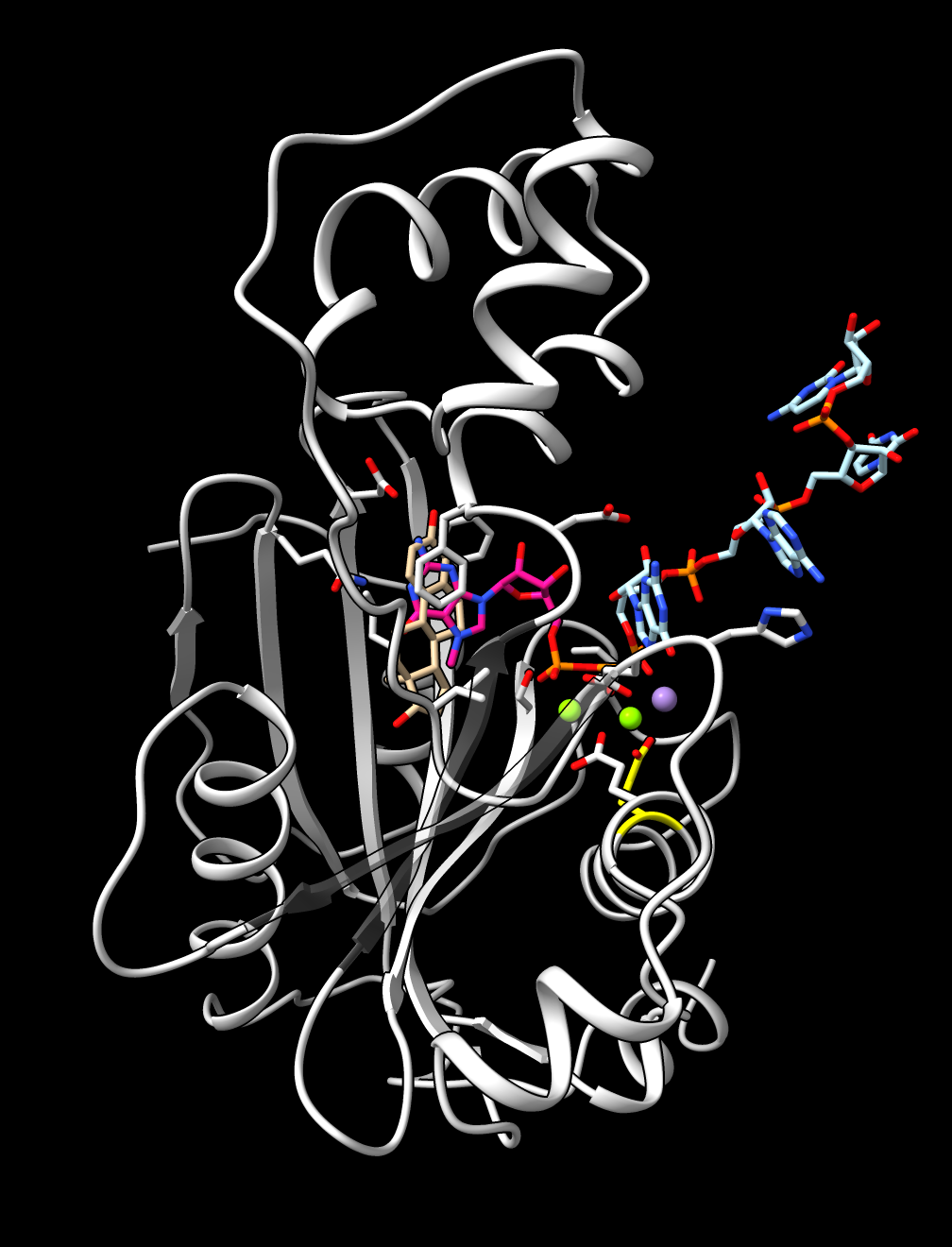
Best scoring FDA ligand estradiol in brown overlaps mRNA cap purine. |
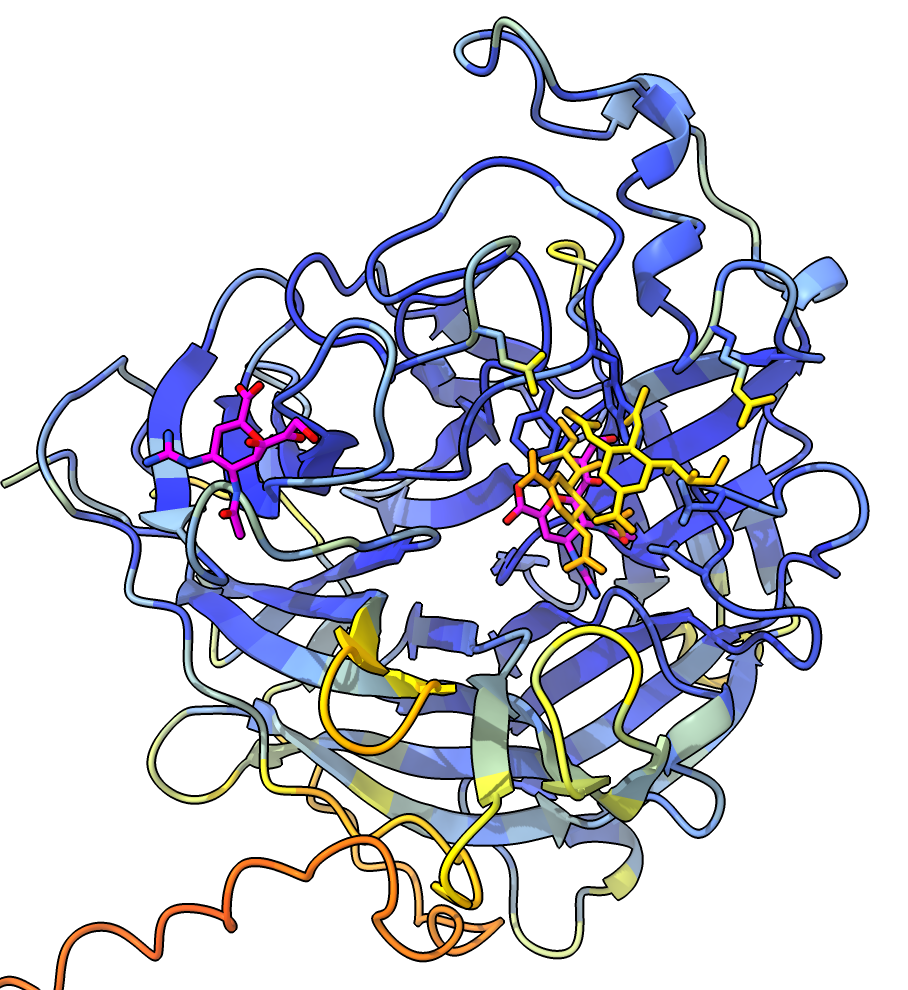
The monkeypox protein we have been calling mpox_d10 has UniProt sequence OPG122. More information about it is available for another pox virus vaccinia UniProt sequence PG122_VACCW where it says it is a decapping enzyme that removes the 5'-cam from host and virual mRNAs, mostly from host mRNAs to shutdown host protein synthesis to minimize immune response. There is no PDB structure of a pox virus decapping enzyme with a capped mRNA but we can predict such a structure. This requires using fancier AlphaFold 3 input that specifies the RNA and covalently links the 7-methylguanosine cap on the 5' end. I used PDB structure 4n48 and cap chemical component dictionary entry MGT to create this AlphaFold 3 input mpox_d10_mrna.json. I used RNA 4-mer GAUC. I added two magnesium and a manganese ions since the vaccinia UniProt page lists those and ligands.
The images at right show this prediction with mRNA in light blue and 5' cap in bright pink with orange triphosphate connecting the two. The ions are positioned between the putative catalytic residue E141 in yellow and the triphosphate where the cap will be cut off. The image on right also superimposes the highest scoring FDA ligand estradiol in tan showing its rings at the same position as the 5' cap purine. The predicted aligned error suggests AlphaFold is confident in this binding prediction, and the ipTM scores are .95 from protein to cap, and .97, .94, .94 from protein to the 3 ions.
The connection I specified in the AlphaFold 3 input file to connect the cap to the mRNA between the second and third phosphate has very bad geometry with the oxygen between those two phosphor atoms being placed badly with short bonds and bad bond angles. This may be a problem of AlphaFold 3 that it cannot make good quality user-specified connections between polymers (the mRNA) and ligands (the cap).
The Alphafold 3 paper says getting the chirality error rate in predictions for ligands was 4.4% in a benchmark. Looking at the top 5 hits for mpox_d10 and comparing to the PubChem structures of the ligands shows AF3 gives the wrong chirality in 2 of 5 ligands (hits 4 and 5 left to right in image, PubChem structures in white, Chimerax session file top_5_ligands.cxs). I wonder if the SMILES strings obtained from PubChem are properly expressing the chirality. Also there are multiple chiral centers for some ligands and probably the 4.4% error rate is for each chiral center.
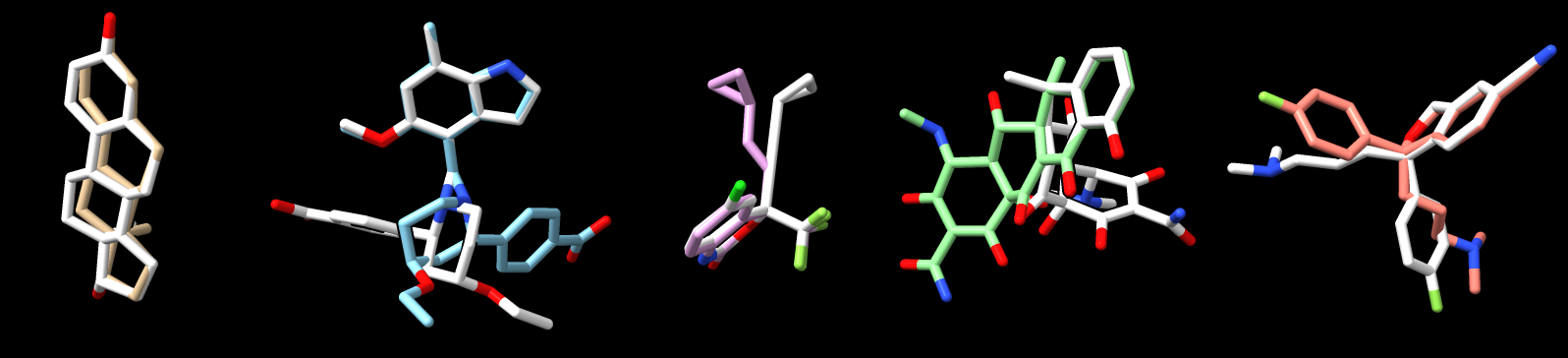
The ligand atom names from PubChem structures differ from the AlphaFold 3 prediction atom names. I looked for chirality inversions by eye after aligning the PubChem and AF3 ligands. To align them I hovered the mouse over corresponding atoms and listed 3 pairs of matching atoms in the ChimeraX align command:
align #6@O2,C13,C7 to #1/D@O2,C12,C8

chlorine atom clash | 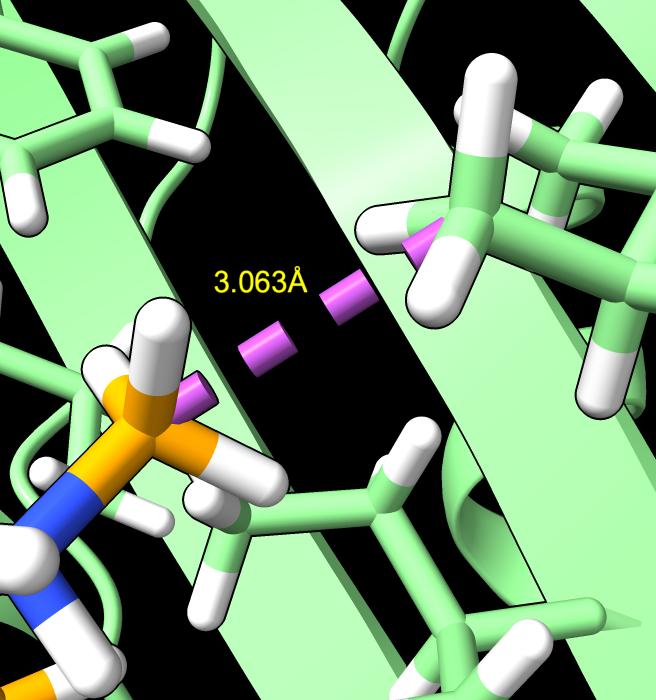
doxycycline (orange) methyl clash |
The ChimeraX clashes command can identify unreasonably short distances between ligand and protein atoms in the predicted structures.
clashes #1/D restrict #1/A reveal true showDist true
Trying this on the top 5 scoring predictions identified clashes in two of them. In iptacopan_hydrocloride the hydrocloride is a separate molecule that AlphaFold places far from the iptacopan and the chlorine atom clashes with several nearby atoms. In the doxycylcine prediction a ligand methyl is about 3 Angstroms from a protein isoleucine methyl where usually methyl-methyl carbon distances are greater than 3.3 Angstroms.
It would be interesting to look at how variable the protein side chain positions in the binding site are across the high confidence docked ligands. In other words, see how much AlphaFold adapts the protein to accomodate the ligand. The simplest approach would be to align all the open predictions and then show all the side-chains with 5 Angstrom of any ligand. One could recognize by eye mobile versus fixed side chain positions.
To quantify the variation in side chain atom positions you could compute the RMSD per-atom. Surprisingly there is not a ChimeraX command for that so I wrote this Python script per_atom_rmsd.py. Opening it in ChimeraX defines the atomrmsd command and "atomrmsd protein" will assign RMSD values to every atom across all the opened structures. You should align the structures first (e.g. "mm #2-9 to #1"). Then the Render by Attribute tool (menu Tools / Depiction / Render by Attribute) could be used to color all atoms by their RMSD, e.g. white for low RMSD and red for high RMSD.