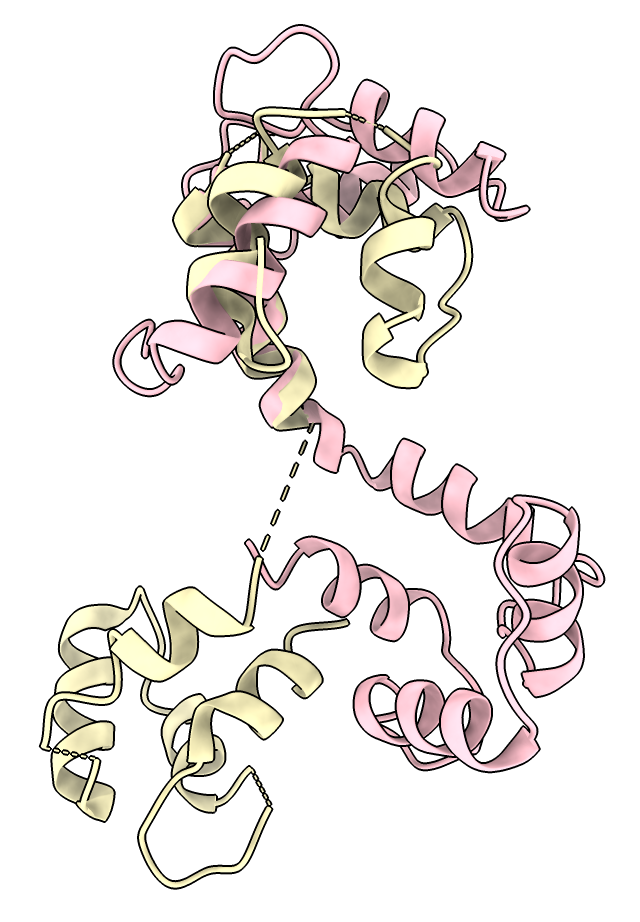

Tom Goddard
August 12, 2024
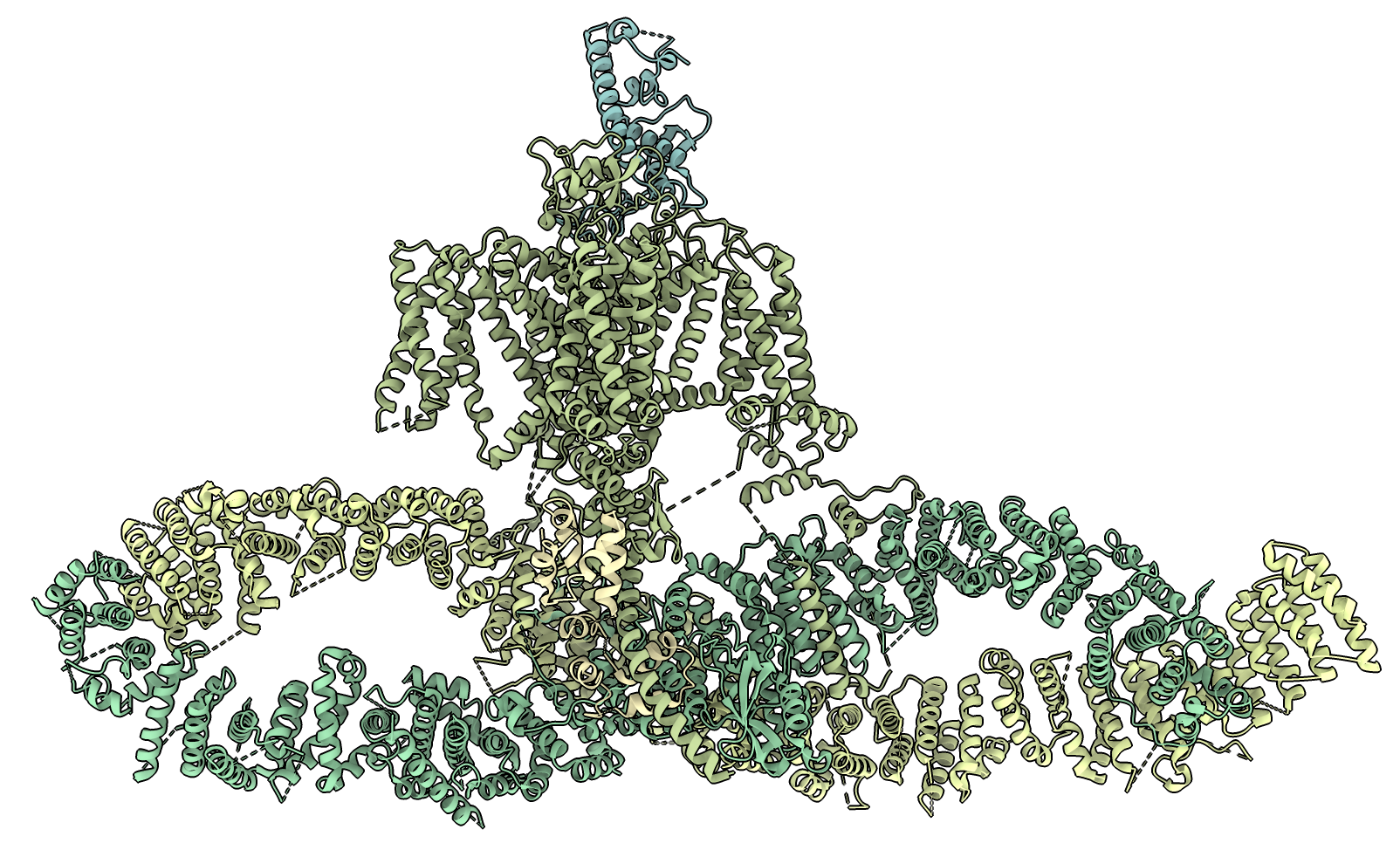
Mike Boucher and Hiten Madhani identified an homolog of human sodium leak channel (NALCN) in Crytpococcus neoformans. The complex consists of 5 proteins and 4 of them have hard to detect sequence similarity. There are 3 human (7sx3, 7sx4, 7wji) and 1 mouse/rat (7w7g) cryoEM structures of the complex, but there are no experimental structures of the Cryptococcus proteins. The putative complex is 7556 amino acids, too large to predict using AlphaFold multimer. The aim here is to predict the 8 protein-protein interfaces seen in the human complex using the Cryptococcus proteins, to support the claim that they form a homologous complex.
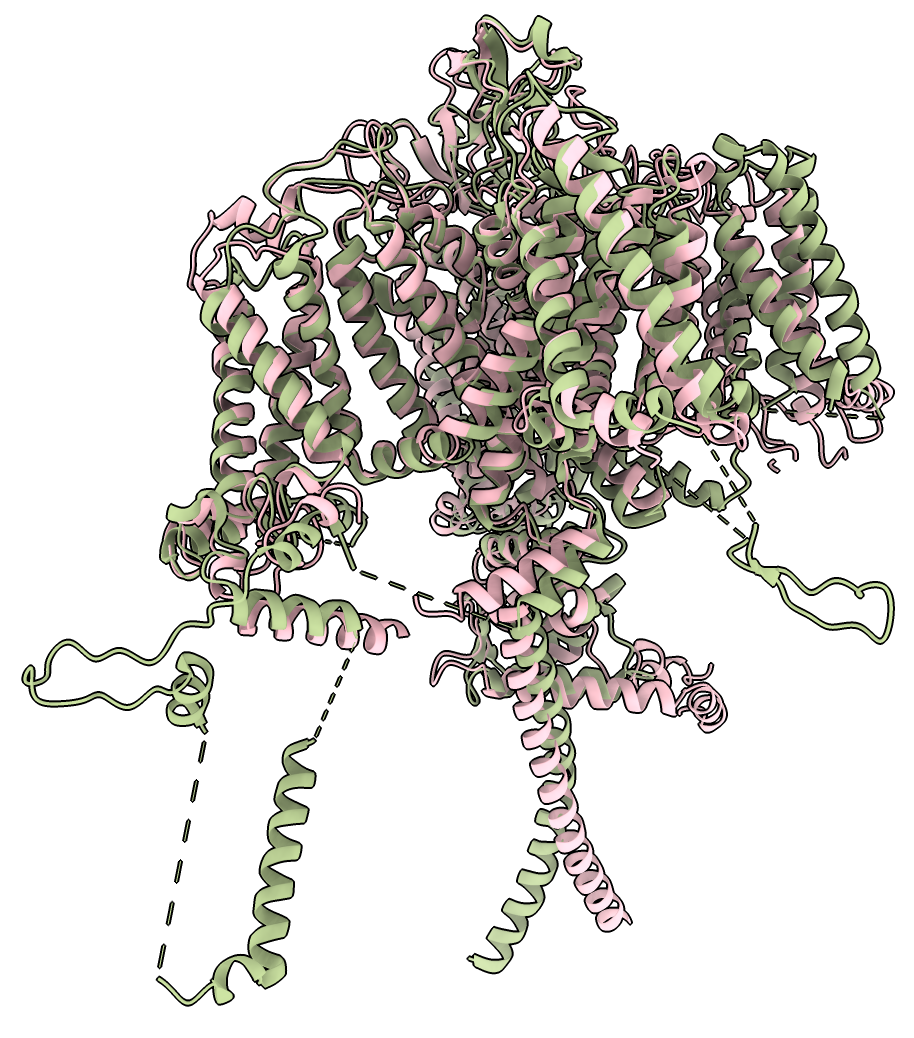
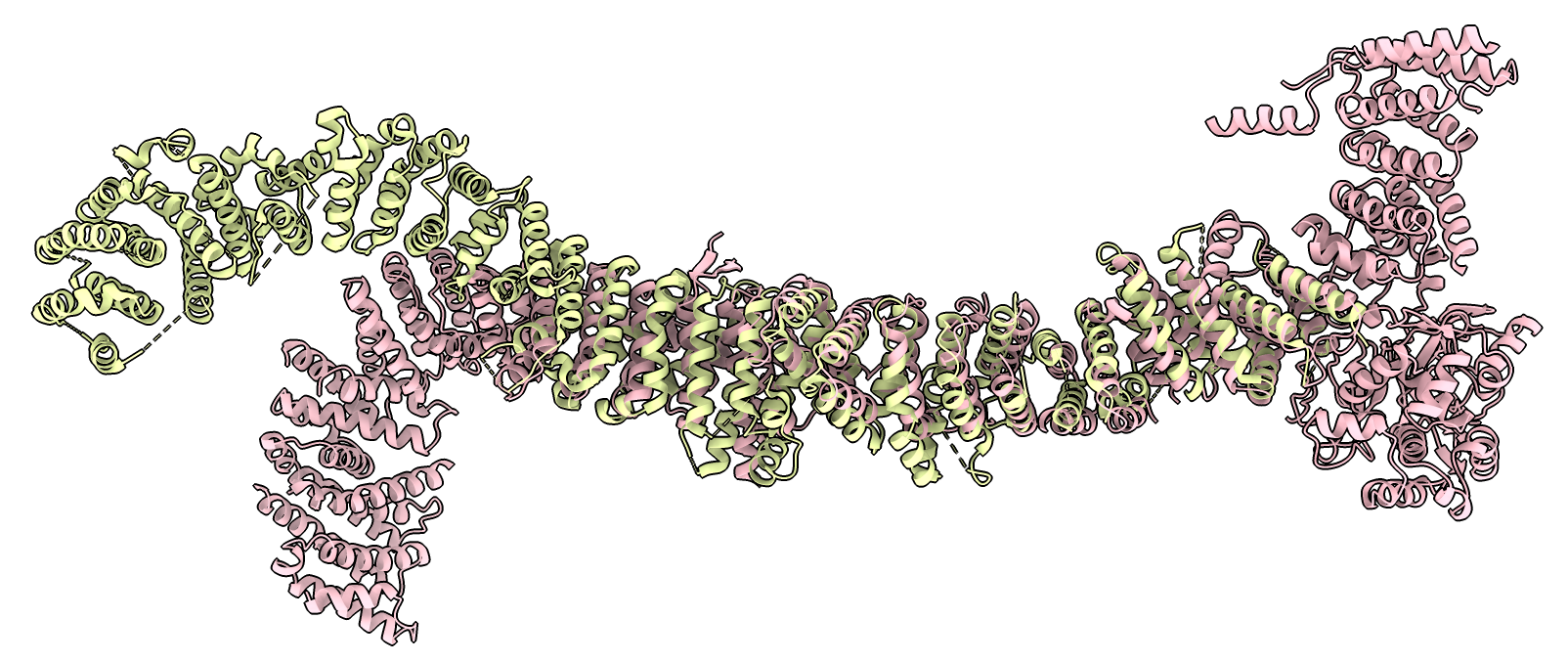
The complex consists of 5 proteins. Compare to human complex PDB 7sx3.
| | | |
| PDB 7sx3 aligned to AlphaFold Cryptococcus predictions (pink) | |||
The sequence identity between Cryptococcus and human is low for 4 of the proteins but Foldseek structure search using the AlphaFold cryptococcus predicted structures finds 3 of the human 7sx3 proteins (CCH1, UNC79, UNC80).
| Human protein | 7sx3 Chain | Length | Crytopcoccus | Length | AlphaFold | Identity | Notes |
|---|---|---|---|---|---|---|---|
| NALCN channel | A | 2042 | CCH1 CNAG_01208 | 2106 | prediction | 22% | Foldseek on alphafold monomer prediction found 7sx3 as best hit (evalue 1e-56). |
| FAM155A transmembrane | B | 483 | MID1 CNAG_03751 | 623 | prediction | Foldseek did not find any low evalue hits for MID1 AlphaFold monomer prediction (min evalue 0.02), nor did it find anything using MID1 from MID1/CCH1 dimer AF2 prediction (min evalue 0.005) or dimer AF3 prediction (min evalue 0.02). But 3 helices of MID1 prediction do align with FAM155A. | |
| Calmodulin-1 | C | 149 | CAM1 CNAG_01557 | 149 | prediction | 83% | Sequence alignment computed using Clustal Omega not Foldseek since seq identity high. Predicted structure does not match 7sx3 well. |
| UNC79 | D | 2561 | CNAG_06362 | 2352 | prediction | 14% | Foldseek using CNAG 01613/06362 dimer prediction finds 7sx3 as 3rd best hit (evalue 1e-14, 7w7g_A 3e-16, 7sx4_D 9e-15). |
| UNC80 homolog | E | 3283 | CNAG_01613 | 2326 | prediction | 14% | Foldseek using CNAG 01613/06362 dimer prediction finds 7sx3 as best hit (evalue 3e-24). |
The sequence identity value in the tables uses only the part of the query sequence that Foldseek aligns and the denominator is the length of that part of the query sequence. Here are the sequences of the Cryptococcus proteins cncalcium_monomers.fasta.
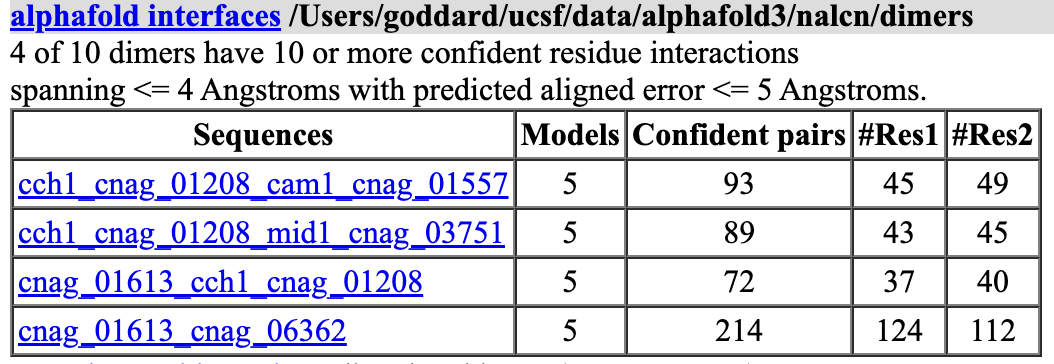
I ran Alphafold 3 of the 5 monomers and 10 possible dimers (excluding homodimers) on Google's server which took only 21 minutes (running all jobs in parallel). I used the ChimeraX alphafold monomers and alphafold dimers commands with the outputJson option to create an AlphaFold 3 input file to setup all the runs. And I used the alphafold interfaces command to tabulate Alphafold confidence of the dimer interfaces.
Alphafold predicts four dimers observed in 7sx3 (CCH1 + CAM1, CCH1 + MID1, CCH1 + 01613, 01613 + 06362) but does not predict two of the 7sx3 dimers (CCH1 to 06362 or CAM1 to 01613). Also it does not give confidence to any dimers not found in 7sx3.
By aligning the AlphaFold predictions for cryptococcus dimers to the 7sx3 structure we can see if the same protein-protein interfaces are present. This is a bit tricky because the sequences of the cryptococcus and human proteins are very different so I need a sequence alignment between them to know which residues correspond to which other residues.
I wrote a new command "ialign" (python code ialign.py) to perform alignments on specified residues using a opened sequence alignment. The idea is to align the AlphaFold dimer models using the interface residues of the 7sx3 structure.
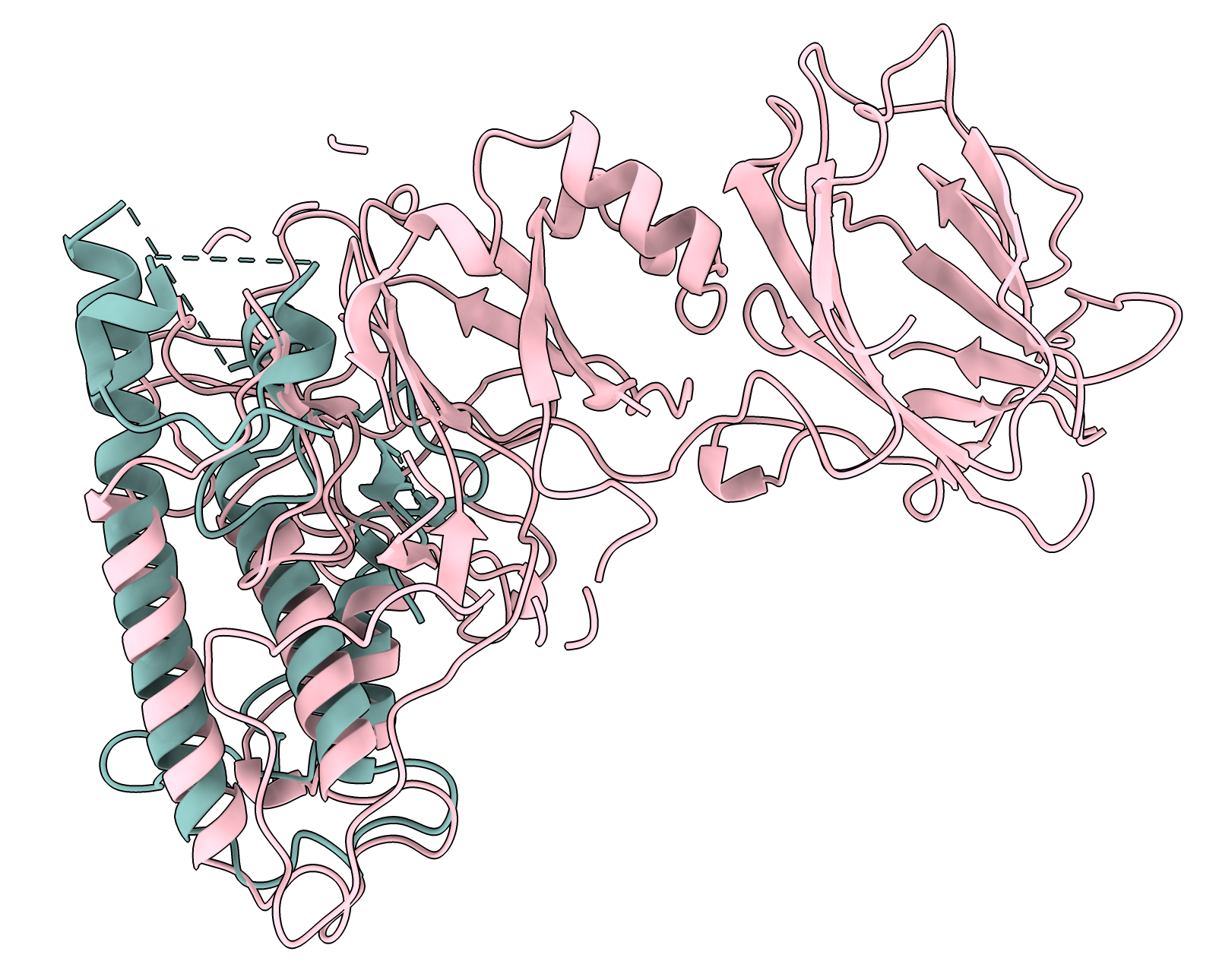
Here is an example where I aligned AlphaFold 3 CCH1/CAM1 dimer prediction to 7sx3 NALCN / Calmodulin-1. We see the two calmodulin domains are packed tightly around the alpha helix in the prediction while they are ~15 A further apart in the human PDB structure. But the prediction does place the calmodulin wrapped around the same NALCN and CCH1 alpha helix. The structure alignment was done on the corresponding CCH1 and NALCN residues with sequence alignment created by Foldseek and used 28 residues of NALCN at the interface with its bound calmodulin. The interface residues were computed by the "patch" command mentioned below.
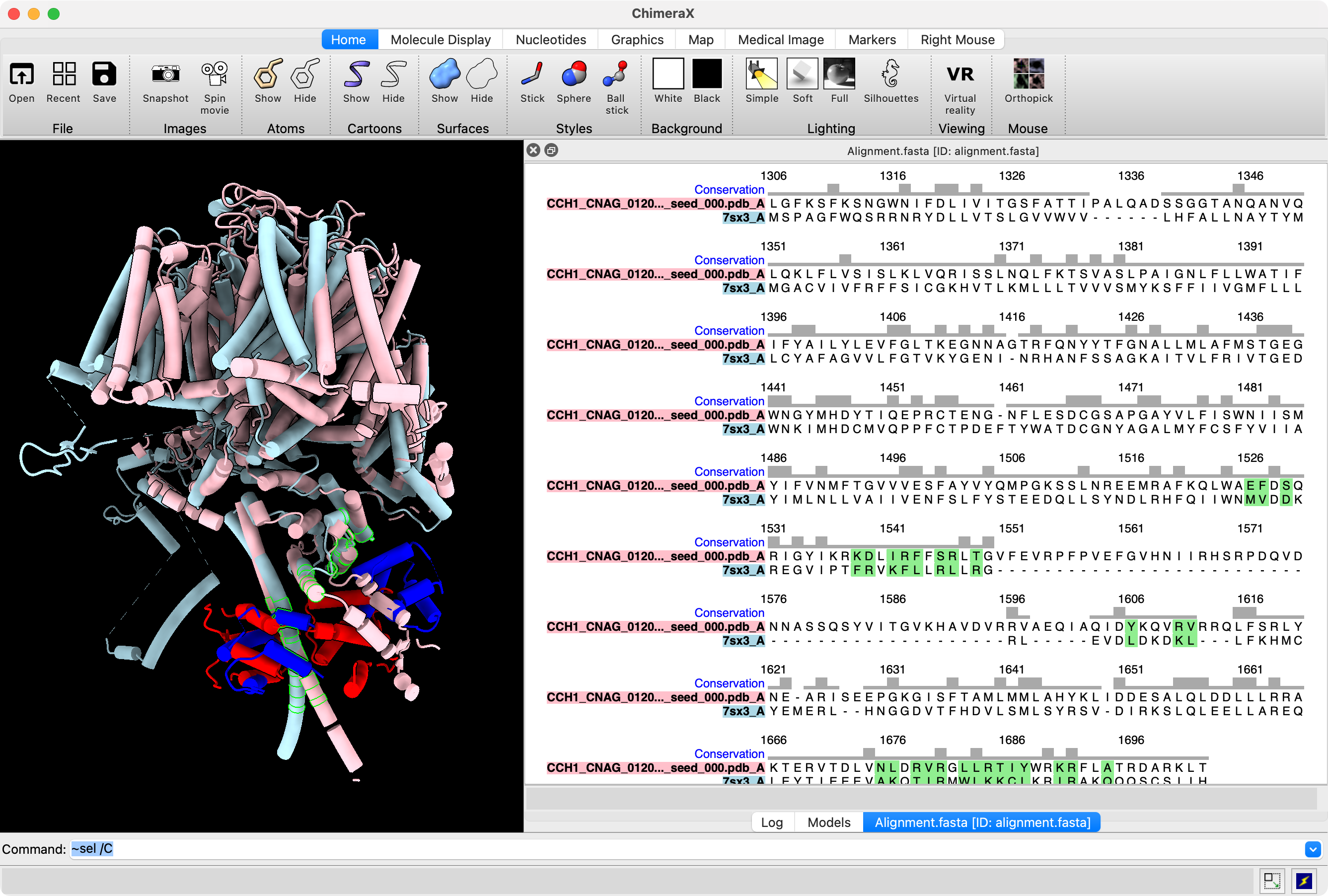
AlphaFold predicted CCH1 (pink) and CAM1 (red) aligned to PDB 7sx3 NALCN (light blue) and Calmodulin-1 (dark blue |
TODO: If we predict dimers of just the portions of cryptococcus proteins near the observed interfaces in 7sx3 will AlphaFold be able to find the interfaces that were not present in the full dimer predictions?

Some new ChimeraX code patches.py implementing the "patches" command reported 10 protein-protein interfaces if human complex 7sx3. Two of the interfaces are small with the channel just touching the UNC79/80 homologs with 3 or 4 residues so we ignore those. The remaining 8 contact patches are: 1) UNC79/80 wrap around each other making 3 interfaces, 2) channel CCH1 contacts the UNC79/80 dimer in two places, 3) CAM1 is sandwiched between CCH1 and UNC80 making two interfaces, 4) CCH1 contacts MID1 in one interface,
D with E, 86 with 88 residues
A with B, 71 with 50 residues
A with E, 42 with 64 residues
A with C, 28 with 31 residues
D with E, 28 with 21 residues
D with E, 24 with 21 residues
A with D, 9 with 17 residues
C with E, 14 with 10 residues
A with D, 4 with 8 residues
A with E, 3 with 4 residues