

Tom Goddard
October 3, 2024
Here is information about the Similar Structures user interface panel and commands. Another page shows a simple example using these capabilities.
The Similar Structures tool is for visualizing hundreds of similar monomeric protein structures. It starts with a query structure and can use Foldseek, MMseqs2 or BLAST to find similar structures from the PDB or AlphaFold datbases. The purpose of the tool is to allow exploring hundreds of structures to derive possibly useful information about the query structure. It can efficiently depict the sequences, backbone traces, clusters of backbone configurations, and ligands for large sets of structures.
These capabilities rely on pairwise sequence alignments between each similar structure and the query structure. The sequence alignments produced by the search method (Foldseek, MMseqs2 and BLAST) are used. The Foldseek method can find very distantly related structures with less than 10% sequence identity by comparing structure folds, while MMseqs2 and BLAST typically align sequences with more than 20% identity.
The Similar Structure tools is found in menu Tools / Structure Analysis / Similar Structures. The same tool is shown by menu entry Tools / Structure Analysis / Foldseek. The two menu entries are used because Foldseek is a widely recognized name.
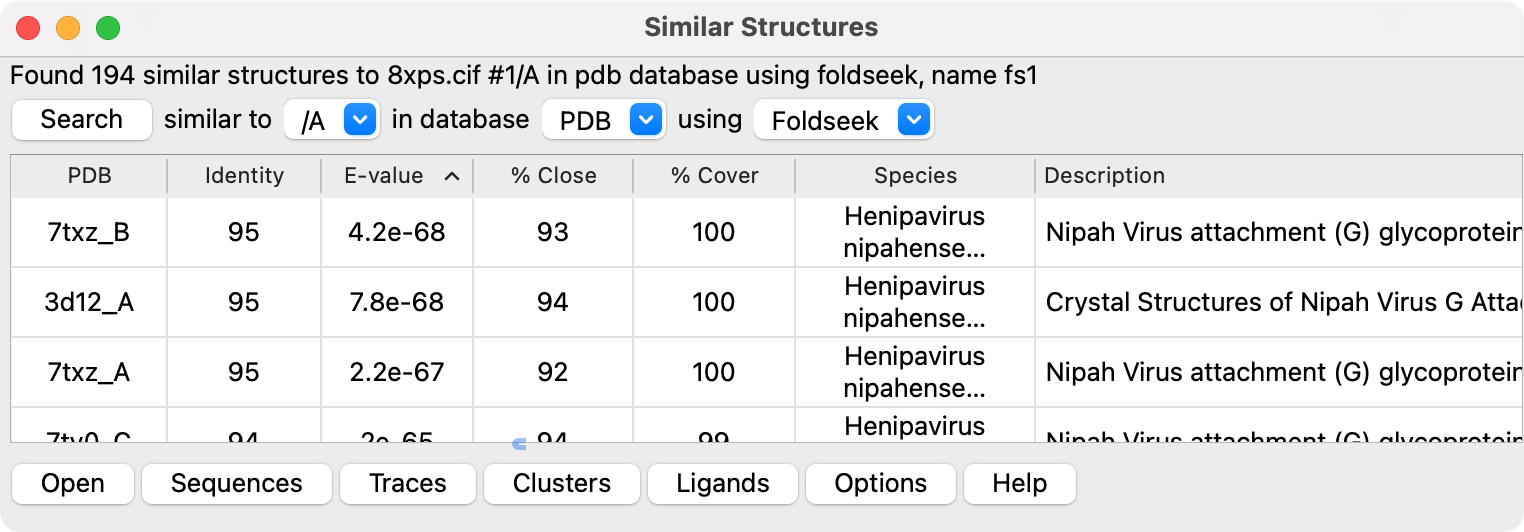
The Search button does a web-based search for similar structures to the specifed chain of an open structure. The tool does not allow searching using only a sequence because most of its capabilities compare the query structure to similar structures. A search using Foldseek uses the Foldseek server from the Martin Steinegger lab (https://search.foldseek.com, hosted in Korea). That Foldseek server uses a clustered version of the PDB database pdb100 which may not be current (the server is currently using a January 1, 2024 version of the PDB as indicated on the server web site). The PDB100 database is clustered and includes only one chain up to 100% sequence identity with at least 95% sequence overlap, reducing about 1 million chains to 340,000. A search using MMseqs2 uses the RCSB search service (https://search.rcsb.org). A search using BLAST uses the UCSF RBVI BLAST web service.
Doing a second search replaces the contents of the first search. Only one set of search results can currently be shown. Search results are saved in ~/Downloads/ChimeraX under subdirectories Foldseek, MMseqs2 or BLAST named using the query structure name with file suffix .sms. This is a JSON file format specific to ChimeraX. The file will be listed in the ChimeraX file history thumbnails so it can easily be reopened without repeating the search.
The % Close and % Cover columns are only reported for Foldseek results since the MMseqs2 and BLAST results don't provide C-alpha coordinates. If the C-alpha coordinates are obtained by fetching the coordinates with the similarstructures fetchcoords command then the table is updated. For sequence-based searches (MMseqs2 and BLAST) a match may have no residues with atom coordinates aligned with the query.
Selecting one or more lines in the table of similar structures and pressing the Open button fetches those structures from the PDB or AlphaFold database and aligns them to the query. The alignment rotates and translates the matched structure to minimize the RMSD of its C-alpha atom to those of the query for paired residues, and uses iterative pruning (default pruning distance 2 Angstroms set in Options). The iterative pruning algorithm does not guarantee that the largest number of residues will be aligned. For multi-domain proteins where the domains are packed differently in the match and the query the algorithm may align either of the domains.
A fetched structure will be trimmed, deleting some residues, based on the trimming options shown when the Options button is clicked. The default trimming options delete


The Sequences button shows an image depicting the sequence alignments of each similar structure to the query. Each row of the image is one sequence, so 200 similar structures would produce an image 200 pixels tall. The columns of the image correspond to the residues of the query structure. The plot primarily shows the portions of the query structure that are covered by the similar structures. For a multi-domain query structure the similar structures may only cover single domains. Hovering the mouse over the sequence plot displays text above the plot indicating the structure for the row the mouse is over, and the query residue number for the column the mouse is over.
The sequence plot shows white where there is no alignment between similar structure and query structure, black where there is an alignment, aqua blue where there is an alignment and the same amino acid type is in the similar structure and query, and yellow at conserved sequence positions where more than 50% of the similar structures that align to a query residue have the same amino acid as the query and 10 or more similar structures are aligned.
Clicking on the sequence plot (left or right mouse button) shows a popup menu that allows loading the structure for the row clicked on, or changing the ordering of the sequences in the plot, or changing the sequence coloring, or coloring the query structure to show alignment properties.

Some menu entries use the structure or query residue at the mouse position where the menu was shown.
The default ordering of the sequences is either e-value or cluster with the latter being used if there are more than 2 clusters.
The initial coloring shows well conserved positions in yellow and positions with same amino acid type as the query in aqua. Both conserved and LDDT coloring can be shown at the same time with the yellow and aqua conservation colors overlaying the LDDT colors.
The query structure residues can be colored using properties derived from the sequence alignment. These colorings use query residue attributes "coverage", "conservation" and "lddt" set when the sequence plot is shown. The "coverage" residue attribute gives the number of similar structures aligned to that residue. The "conservation" attribute gives fraction (0-1) of structures aligned at that residue having the most common amino acid type at that position. The most common amino acid type may not be the type in the query structure. The "lddt" attribute gives the local distance difference test score that measures how similar the structure is to the query in a neighborhood of 15 Angstroms of the query residue C-alpha atom. It only compares the aligned residues of the similar structure and query.
|
|
|
|
The traces button creates a single model showing C-alpha backbone traces for all of the similar structures aligned to the query structure. The purpose is to see what parts of the query are covered and how well the similar structures match the query. Soft lighting improves the appearance of the traces hairball. Backbone trace models are saved in sessions.
The traces are put in a single atomic model containing only backbone C-alpha atoms with each similar structure contributing one chain with chain identifier equal to the database id of the similar structure. The C-alpha atoms have the residue type of the similar structure but use the residue numbers of the query structure. They do not have secondary structure assignments (alpha helix, beta sheet). Foldseek search results include the C-alpha coordinates so it is not necessary to fetch the database structures to show the backbone traces. For mmseqs2 and blast search results the the database structures will be downloaded to obtain the C-alpha coordinates.
By default trace ribbons are shown for similar structure C-alpha atoms within 4 Angstroms of the corresponding query C-alpha atoms, as well as contiguous segments of at least 5 residues that have maximum distance 10 Angstroms to the query. These heuristic rules limit the trace display to parts that are close to the query. Different parameters to restrict trace display can be applied with the similarstructures traces command.
Using ctrl-double-click on a trace ribbon or atom shows a context menu that allows fetching and opening the full atomic model represented by that trace, or showing its row in the similar structures table, or showing only the ribbon for that similar structure, or showing ribbons for all traces, or showing full length traces or only the parts close to the query.

The Clusters button attempts to cluster the backbone traces of the similar structures based on their shapes. The structures are plotted in 2 dimensions with each similar structure shown as a circle with the name of the structure in small text. Structures with similar backbone paths are plotted close together and clusters in the plot are colored. The colors can be applied to the backbone traces using the popup menu on the cluster plot. Cluster plots are saved in ChimeraX session files (suffix .cxs).
The layout of structures in the plot works by choosing a small number of query residues. It concatenates the C-alpha atom coordinates (x,y,z) of each similar structure C-alpha atom that aligns with a chosen query residue producing a vector of length 3*N for N chosen residues. That vector is a spatial summary of the backbone that is projected to 2 dimensions by the UMAP (Uniform Manifold Approximation and Projection) algorithm. Those plotted points for each structure are clustered by distance and the clusters are assigned random colors. The Cluster button chooses the 5 most conserved sequence positions (using the conservation residue attribute). Structures which don't have residues aligned for all 5 are not plotted or clustered.
Clicking on the cluster plot shows a menu. If the click is on a plotted structure point then some actions specific to the cluster that structure belongs to are included.

| 
|
To color backbone traces or change cluster colors:
To see more details about a particular structure:
To see where the chosen query residue C-alpha atoms are located:

| 
|
The ligands button copies the ligands from the similar structures onto corresponding locations on the query structure. It makes a single new atomic model consisting of all the ligands from the similar structures. This requires fetching and opening all the structures which can take several minutes for a few hundred structures with a fast network connection. The log output reports what ligands were copied, for example,
Found 16813 ligands in 165 hits: 6Y6, 8LM, ACT, CA, CIT, CL, DAN, DF4, EDO, EPE, FSI, GBL, GLC, GOL, HOH, IMD, IPA, K, MAN, MG, MPD, NA, NAG, NDG, OEL, PEG, PO4, SFJ, SO4, YT3, ZMR, ZN
Each non-polymer residue of a similar structure is evaluated to see if it can be mapped on to the query structure as follows. All protein residues in the structure within 5 Angstroms of the ligand are found and at least half of those must be aligned to query structure residues. If a minimum RMSD alignment between those neighboring residues and the corresponding query residues has RMSD less than 3 Angstroms then the ligand is transferred to that position aligned to the query. Other ligands are discarded. Different parameters can be used with the similarstructures ligands command.
Often thousands of water molecules, and ions, and crystallization adjuvants are found and they can be hidden to get a better view of more interesting ligands with ChimeraX commands
hide solvent
hide ions
hide :SO4
Hovering the mouse over a ligand of interest pops up a description showing the source structure and ligand 3-letter code. That structure can then be loaded using the table of similar structures (sorting on name to find the structure of interest).
The Options button displays a few options at the bottom of the Similar Structures panel. Clicking the Options button again hides the panel.
The trim options effect the behavior of the Open button and cause it to delete certain parts of the fetched and opened structures as described in the section describing Open.
The alignment pruning option provides a distance cutoff (default 2 Angstroms) used when aligning similar structures to the query structure. C-alpha atoms in the structure further than this distance from their corresponding atom in the query after initial alignment are not used in subsequent alignment interations. This is used for alignment when opening a similar structure, and also used for aligning backbone traces, and for clustering backbone shapes, and for copying ligands. For more details of how the iterative pruning is done to determine which residues are aligned see the ChimeraX align command.
The Traces, Clusters, and Ligands buttons by default act on all similar structures in the table. To apply those capabilities to just a selected set of similar structures enable the Traces, clusters and ligands for selected rows only option. The sequences plot always applies to all structures in the table.
The following commands can be used as an alternative to the graphical user interface for working with similar structures. The commands in many cases offer more settings to adjust the behavior.
foldseek chain-spec [database pdb100|afdb50|afdb-proteome|afdb-swissprot] [trim true|false|chains,sequence,ligands]
[alignmentCutoffDistance dist] [saveDirectory dir] [wait true|false] [showTable true|false]
- Search for proteins with similar folds using Foldseek web service.
Defaults: saveDirectory ~/Downloads/ChimeraX/Foldseek.
The foldseek command uses the Foldseek server (https://search.foldseek.com/search) to find structures with similar topology. It can search clustered versions of the PDB database (pdb100) or AlphaFold database. Only a representative structure is included for each cluster. The clustering is described in the Foldseek publications (e.g. PDB is 100% sequence identity and at least 95% coverage). The current pdb100 database used by the server was created January 1, 2024 so does not contain recent structures. The server does not have e-value or maximum number hit parameters but currently has a maximum hit limit of 1000. The trimming and alignment cutoff distance options are not used in the search but set the default values when structures are later opened with the similarstructures open command or via the table user interface. The results are automatically saved in a ChimeraX-only similar structure file format (suffix .sms). This allows reopening the results in later ChimeraX sessions without rerunning the search. The saved file is listed in the ChimeraX file history. The result tables are also saved in ChimeraX sessions. By default the search is done in the background (wait false) so ChimeraX can still be used while waiting for search results. The search typically takes tens of seconds. The results are usually shown in a table (showTable true), but the search can be done without creating a table. Search results are given a name reported in the log such as "fs1", "fs2", ... and these search result names can be used in analysis commands. Currently there is only one displayed table and a search replaces any current results in the table.
sequence search chain-spec [database pdb|afdb] [evalueCutoff evalue] [identityCutoff identity] [maxHits N]
[trim true|false|chains,sequence,ligands] [alignmentCutoffDistance dist] [saveDirectory dir] [showTable true|false]
- Search for proteins with similar sequences using RCSB MMseqs2 web service.
Defaults: evalue 1e-3, identityCutoff 0, maxHits 1000, saveDirectory ~/Downloads/ChimeraX/MMseqs2.
The sequence search command uses the RCSB search API (https://search.rcsb.org/) to perform sequence searches of experimental or AlphaFold models. Although the RCSB search API does not specify the sequence search algorithm it is currenlty using MMseqs2 which is much faster than BLAST at similar sensitivity (reported to be 100-fold faster in the MMseqs2 paper). It takes about 5 seconds to search the PDB and about 30 seconds to search the 200 million predicted structure AlphaFold database. ChimeraX is frozen until the search completes. If the showTable option is false then no table of results is shown but other similarstructures commands can still be run on the results.
similarstructures blast chain-spec [database pdb|afdb] [evalueCutoff evalue] [maxHits N]
[trim true|false|chains,sequence,ligands] [alignmentCutoffDistance dist] [saveDirectory dir] [showTable true|false]
- Search for proteins with similar sequences using UCSF RBVI BLAST web service
Defaults: evalue 1e-3, maxHits 1000, saveDirectory ~/Downloads/ChimeraX/BLAST.
The similarstructures blast command is similar to the ChimeraX blastprotein command but produces a similar structures table of results focused on structure comparison while blastprotein produces different table focused on displaying additional attributes of the structures harvested from the databases. Both of these BLAST searches use the UC San Francisco Resource for Biocomputing, Visualization and Informatics (UCSF RBVI) BLAST server. The server PDB sequence database is updated weekly. AlphaFold database searches are slower than MMseqs2 taking about 5-10 minutes. ChimeraX is frozen when using the similarstructures blast command until the search completes. If the showTable option is false then no table of results is shown but other similarstructures commands can still be run on the results.
similarstructures fromblast [blast-name] [save true|false] [saveDirectory dir]
- Make a similar structures table from currently shown blast results
Defaults: saveDirectory ~/Downloads/ChimeraX/BLAST.
The similarstructures fromblast command displays BLAST search results made with the blastprotein command in a similar structures table. The blastprotein command has to be run on a chain of an open structure rather than a sequence without structure to use the similar structures capabilities. If more than one set of blast search results are open the name of the blast results (listed in the title bar) can be specified.
similarstructures open struct-name [trim true|false|chains,sequence,ligands] [align true|false] [alignmentCutoffDistance dist]
[inFileHistory true|false] [log true|false] [fromSet set-name]
- Open a structure from similar structure search results and align it to the query structure.
This command fetches the a structure from the search results. The structure name is the database identifier, for the PDB it has the accession code followed by an underscore and chain identifier, e.g. 6cmi_B. For the AlphaFold database it is a UniProt accession code. Then name should be exactly as it is shown in the table of search results including case. Parts of the database entry which are not aligned to the query may be deleted depending on the trim settings as described above. By default the opened structure will be aligned to the query using the sequence alignment provided by the search method. Iterative pruning of the aligned pairs or residues is done the same as with the ChimeraX align command. When opening large numbers of structures it may be desirable to not include them in the file history (option inFileHistory false) or describe them in the log (option "log false").
similarstructures sequences [showConserved true|false] [conservedThreshold frac] [conservedColor a color]
[identityColor a color] [lddtColoring true|false] [order cluster|evalue|identity|lddt] [fromSet set-name]
- Show an image of all aligned sequences from a similar structure search, one sequence per image row.
Defaults: conservedThreshold 0.5, conservedColor 225,190,106, identity_color 64,176,166, order cluster or evalue if only one cluster.
The sequence plot is described above.
similarstructures traces [alignWith align-residues] [alignmentCutoffDistance dist]
[show all | close] [distance dist] [maxSegmentDistance sdist] [minSegmentResidues N] [breakSegmentDistance bdist]
[fromSet set-name] [ofStructures struct-names] [replace true|false]
- Show backbone traces of similar structures aligned to query structure.
Defaults: alignWith all aligned residues with pruning, distance 4.0, maxSegmentDistance 10.0, minSegmentResidues 5, breakSegmentDistance 5.0.
This command displays backbone traces using C-alpha atom only chains for each similar structure as described above. Each backbone trace is a separate chain with chain identifier equal to the similar structure database id. The alignWith option limits structure alignment of similar structure to use only the specified query residues. This for instance could allow aligning a specific domain if the query has multiple domains. The options show, distance, maxSegmentDistance, minSegmentResidues, and breakSegmentDistance can limit which residues are initially shown in the trace to those close to the query structure. The show all option will show all the residues and ignore the other four options. The default show close limits the display to just C-alpha atoms within the specified distance value of the paired query C-alpha plus segments of consecutive residues that don't get far away. The ends of segments are defined by places where two consecutive C-alphas in the hit are greater than breakSegmentDistance (default 5 Angstroms) away from each other. Foldseek results do not include coordinates or sequences for insertions in the hits that don't align with the query, so this break distance test serves to detect where there is likely a missing insertion. A segment is displayed if it is at least minSegmentResidues in length and its maximum C-alpha distance to paired query C-alpha is less than maxSegmentDistance. C-alpha atoms that don't meet the closeness criteria will be part of the backbone trace model but their ribbon will not be shown unless the user runs separate commands to show them (e.g. "show #2 ribbons").
similarstructures cluster [query-residues] [alignWith align-residues] [alignmentCutoffDistance dist]
[clusterCount N] [clusterDistance cdist] [colorBySpecies true|false]
[replace true|false] [fromSet set-name] [ofStructures struct-names]
- Show plot of similar structure backbone conformation clusters computed using specified query residues.
Defaults: alignWith all aligned residues with pruning, clusterCount not used if not specified, clusterDistance not used if not specified.
The cluster calculation and plotting is described above. The coordinates of C-alpha atoms paired with the specified query residue C-alpha atoms are used to form a vector of length N*3 and these vectors are projected to two dimensions using the UMAP algorithm. When this command is first run the large UMAP Python package umap-learn is installed with ChimeraX and this may take a minute. If a large number of query residues are specified (e.g. the default is to use all query residues) then there may be no similar structures which have all those residues and then there will be no structures to cluster. So it is important to choose a small set (1 - 30) of residues so that many structures will have a complete set of aligned residues. For example, the residues near a binding site would be a reasonable choice to find different binding site geometries. After the UMAP projection there are two methods for deciding how to group the structures into clusters. No grouping is done unless the clusterDistance or clusterCount options is specified. If clusterDistance is given (e.g. 1.5 Angstroms) then projected structures will be grouped that lie with that distance of each other. In other words that is minimum separation of two clusters. If clusterCount (e.g. 5) is given then the k-means algorithm is used to produce that number of clusters. Only one of the two options should be given. If a cluster plot is already shown it will be reused to show the new clustering unless the "relpace false" option is given in which case a new plot window will be created.
similarstructures ligands [rmsdCutoff rmsd] [alignmentRange dist] [minimumPaired frac] [combine true|false]
[fromSet set-name] [ofStructures struct-names] [warn true|false]
- Copy similar structure ligands to the query structure.
Defaults: rmsdCutoff 3.0, alignmentRange 5.0, minimumPaired 0.5.
Mapping ligands from similar structures onto the query structure was described above. The rmsdCutoff, alignmentRange and minimumPaired parameters control how similarly located the residues near the ligand must be to map the ligand onto the query structure. The combine option merges all found ligands from the similar structures into a single model for convenience, and this model can be saved in mmCIF format if desired (menu File / Save or command "save ligands.cif model #3"). When the ligands are merged into a single model the PDB id and chain id of the structure the ligand came from are combined to form the chain identifier in the merged model (e.g. chain id "2cml_B"). This allows discovering where the ligands originally came from. The "combine false" option does not merge the ligand models. In that case a separate model for each similar structure that has mappable ligands is created that contains all the mapped ligands and the chain identifiers and not redefined. If the warn option is true then the command will warn the user that it may take several minutes to download the structures and allow them to cancel the action.
similarstructures fetchcoords [minAlignedCoords N] [ask true|false] [rewriteSmsFile true|false] [updateTable true|false]
[fromSet set-name] [ofStructures struct-names]
- Fetch C-alpha coordinates of similar structures for clustering and showing backbone traces
Defaults: minAlignedCorods 10.
Search results from Foldseek include the C-alpha coordinates of the similar structures which allows showing backbone traces and clustering without fetching the structure models. MMseqs2 and BLAST search results do not include the C-alpha coordinates so to use traces or clustering the structures must be fetched. The fetchcoords subcommand fetches the structures from the database which are then cached in the ~/Downloads/ChimeraX directory and reads the C-alpha coordinates. If the rewriteSmsFile is true then the coordinates will be written into the file with the saved search results for future use. If updateTable is true then the displayed search results table will add columns for % coverage and % close. The "ask true" option shows a dialog asking if you want to fetch the structures before proceeding because this can take many minutes during which ChimeraX will be frozen. The "ask true" option is used when you request traces or clusters and the coordinates need to be fetched.
similarstructures scrollto struct-name [fromSet set-name]
- Show table row for a similar structure.
This command scrolls the search results table to the line with the named structure (e.g. 6cmi_B) and selects that line.
similarstructures pairing struct-spec [color a color] [radius r] [halfbondColoring true|false] [fromSet set-name]
- Show pseudobonds between paired residues for a similar structure search result and the query structure
Defaults: color gold 255,215,0, radius 0.1, halfbondColoring false.
This command helps visualize the pairing of residues between a similar structure and the query by creating pseudobonds (lines) between paired residues. The first argument is the id number of the similar structure which must already be opened. The sequences alignments returned by Foldseek can have very low sequence similarity (5-10%) so visualizing the pairing on the structure can be more revealing than looking at the two aligned sequences.
similarstructures seqalign struct-spec [fromSet set-name]
- Show sequence alignment from a similar structures search for one hit
This command shows the sequence alignment provided by the search method between one similar structure and the query in a standard ChimeraX sequence viewer window. This is another way to inspect the alignment.
similarstructures list - List the names of open sets of similar structures
This command lists the names of the currently open similar structure sets. Names for foldseek searches will be fs1, fs2, ..., MMseqs2 searches will be mm1, mm2, ..., and BLAST searches bl1, bl2, .... These identifiers can be used in the above commands as the fromSet option to specify which set the command applies to. Currently only one set can be displayed in a grpahical table, but other sets can be opened with the showTable false of the foldseek command.
similarstructures close set-name - Close a set of similar structure search results
This command closes an open set of similar structure search results. If the results are shown in a graphical table then that table is closed. If they are not shown the set is closed and will not be saved in sessions.
trim from GUI options otherwise True. alignmentCutoffDistance from GUI options otherwise 2.0. fromSet currently similar structures set shown in GUI panel. ofStructures all similar structures.
This ChimeraX-only file format is a JSON text file that contains search results including the path to the query structure file, the database identifiers of similar structures, sequence alignments to the query, and in some cases C-alpha coordinates of the similar structures so that backbone traces and clustering can be done without loading all of the structure files from the database. The similarstructures fetchcoords command can add these C-alpha coordinates to the file for MMseqs2 and BLAST search results. These files can be opened by ChimeraX creating the table of structures and it will associate an already open query structure if one is found, or try to load the original query structure from the file path contained in the .sms file.