

 home
overview
research
resources
outreach & training
outreach & training
visitors center
visitors center
search
search
home
overview
research
resources
outreach & training
outreach & training
visitors center
visitors center
search
search
home
overview
research
resources
outreach & training
outreach & training
visitors center
visitors center
search
search
home
overview
research
resources
outreach & training
outreach & training
visitors center
visitors center
search
search

Andrej Sali¹, Tom Goddard², Greg Couch², and Conrad Huang²
¹
California Institute for Quantitative Biometical Research (QB3)
University of California, San Francisco
²
Resource for Biocomputing, Visualization, and Informatics
University of California, San Francisco
Background
The Sali group at UCSF (http://salilab.org) is using computation grounded in the laws of physics and evolution to study the structure and function of proteins. We aim to improve and apply methods for: (i) predicting the structures of proteins; (ii) determining the structures of macromolecular assemblies; (iii) annotating the functions of proteins using their structures. Most of our methods are implemented in our software package MODELLER (http://salilab.org/modeller). This research contributes to structure-based functional annotation of proteins and thus enhances the impact of genome sequencing, structural genomics, and functional genomics on biology and medicine. The visualization by CHIMERA of sequences, structures, and alignments of individual proteins and their assemblies is a key tool in the development of our methods as well as in the analysis and presentation of our results. The powerful capabilities of CHIMERA are especially helpful for complex manipulations that include additional information, such as dynamic trajectories and mass density maps from cryo-electron microscopy. CHIMERA is our primary molecular visualization package. More recently, we began to collaborate with Tom Ferrin and Wah Chiu (Baylor College) to integrate the EMAN package for cryo-electron microscopy data processing, CHIMERA for visualization, and our program MODELLER for model building of sequences restrained by related known structures and electron microscopy maps. The integrated system will greatly increase the productivity of electron microscopists as well as the quality of their results. We further illustrate our interactions with the RBVI by more detailed descriptions of four specific collaborations.
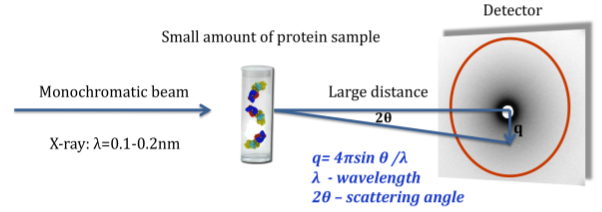
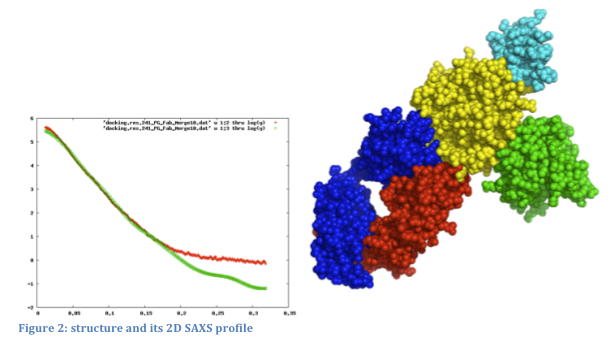
Structural refinement and fitting using Small Angle X-Ray Scattering (SAXS) data
A SAXS measurement determines rotationally averaged scattering intensity of a molecule as a function of spatial frequency (SAXS profile), typically at 1 to 3 nm resolution. The experiment is simple and typically takes several hours. In recent years, SAXS experiments have become increasingly popular, and computational tools for data interpretation are required (Petoukhov and Svergun, 2007; Putnam, et al., 2007).SAXS profiles can be very useful in modeling assembly configurations when the structures of the individual component proteins are known. Given an assembly model, a SAXS profile can be computed and compared to the experimental one. We can further use SAXS profiles for scoring and refinement of alternative assembly models. Currently, there is no graphical interface that ties a 3D structure to its corresponding 2D SAXS profile: structures and profiles are viewed separately in molecular viewers and plotting tools, respectively.
We will support the use of experimental and computed SAXS profiles to guide the modification and refinement of modeled structures (Forster, et al., 2008; Krukenberg, et al., 2008). The following ways of combining 3D structure and SAXS profiles will be implemented: 1) loading a structure and SAXS profile simultaneously and displaying them together; 2) computing and displaying the SAXS profile for part or all of the displayed structure; 3) fitting the computational SAXS profile to the experimental one and displaying them together; 4) recalculating the SAXS profile automatically as the structure is modified (for example, by changing torsion angles or moving proteins relative to each other); and 5) refining the assembly model to better fit the experimental SAXS profile using an IMP web service.
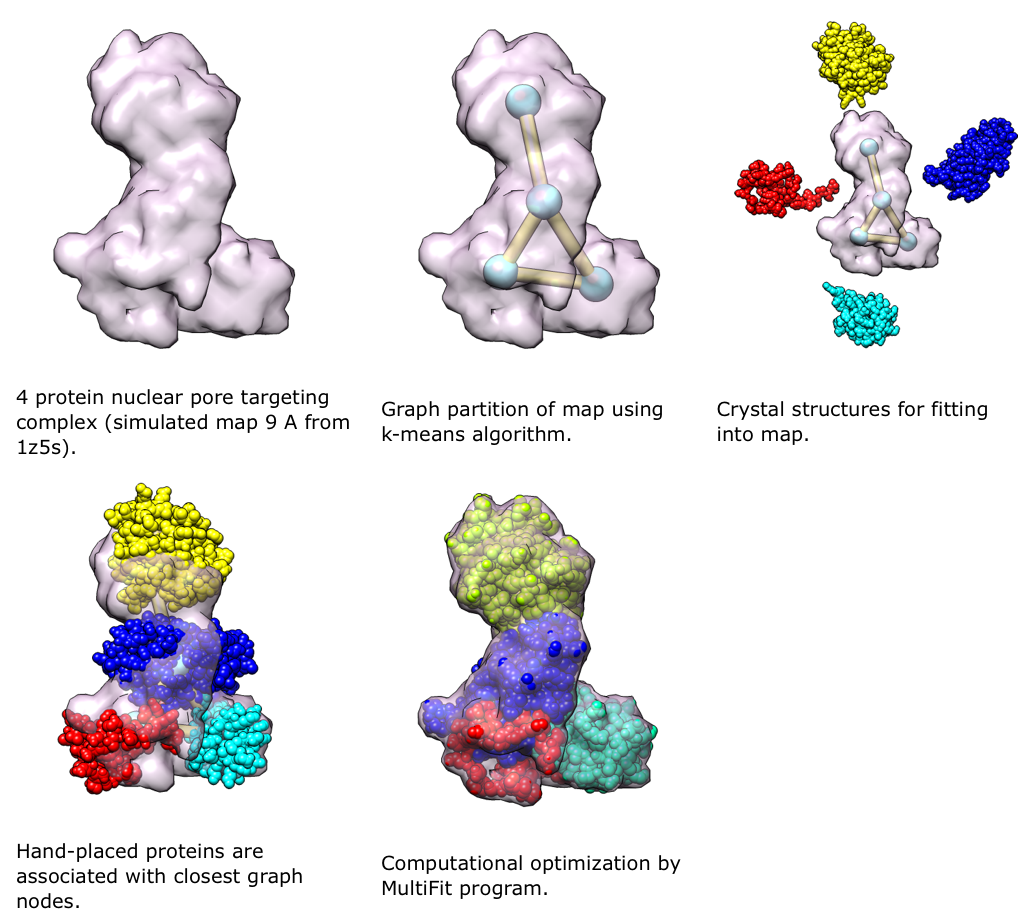
Determination of the structures of large assemblies using cryoEM and proteomics data.
MultiFit (Lasker, et al., 2009) is a new method in IMP for determining the configuration of multiple high-resolution protein structures in an assembly, given a density map of the assembly. It uses the quality-of-fit of each protein in the density map, the protrusion of each protein from the map envelope, and the shape complementarity between pairs of proteins. The combination of these terms reduces the ambiguity of the final solution compared to that obtained with any individual term. Proteomics data can be included in MultiFit, both as additional restraints in the scoring function and to target the sampling.We will use Chimera to view and guide the process of determining the structures of large assemblies with IMP using cryoEM and proteomics data. The user will be able to view and refine models at intermediate steps of the optimization process. Chimera will be used to display the density map and an anchor graph, which shows the approximate positions of protein centroids in the assembly and the interactions among them. The anchor graph is calculated by the MultiFit. We will support the following ways of interacting with the MultiFit process: 1) manual positioning and orientation of the proteins in the density guided by the anchor graph and then calling MultiFit for local refinement, 2) manual positioning of proteins on anchor points but using MultiFit to search all possible orientations, 3) manual positioning and orientation of proteins within the assembly model guided by dynamic feedback of the individual restraint values and overall score from MultiFit, and 4) running MultiFit without any initial positioning but allowing manual refinement at intermediate steps as described above.
CHIMERA for Displaying the Contents of MODBASE
MODBASE is a comprehensive database of annotated comparative protein structure models for all known protein sequences that are detectably related to at least one known protein structure [1,2]. The database is freely accessible to the academic community through a web-interface at (http://salilab.org/modbase). MODBASE contains ~3 million models for domains of 1.3 million unique protein sequences, in addition to the corresponding fold assignments, sequence-structure alignments, model assessments, information about putative ligand binding sites, and single point mutations. MODBASE is bidirectionally linked with a variety of major biological databases, including Uniprot at EBI and Human Genome Browser at UCSC. We collaborated with the RBVI to allow users of MODBASE to visualize the alignments and annotated models directly from the MODBASE interface [1]. To achieve this goal, we created an extension to CHIMERA. The data contained in a MODBASE entry are divided among three different files: a template structure file, a model file, and an alignment file. Manually downloading and opening these files with visualization tools can be a cumbersome process. The new CHIMERA extension enables a web browser to communicate directly with CHIMERA. Clicking on a single link associated with each MODBASE model triggers CHIMERA to start on your local computer. Information related to the model is transmitted to CHIMERA via a registered MIME (Multipurpose Internet Mail Extensions) file type, which then displays the structures of the template and the model; their alignment is also displayed using CHIMERA's multiple sequence alignment viewer, MultAlign Viewer. The user can then apply CHIMER's rich set of visualization and analysis tools to further study the model. Additionally, for models with associated point mutations and putative ligand binding sites, the relevant residues are automatically highlighted in CHIMERA. In the future, MODBASE will contain models based on multiple templates. We plan to adapt the MODBASE-CHIMERA interface to display the complex multiple alignments in a most user-friendly way. CHIMERA as a Graphical Interface in the Modeling Process
CHIMERA has recently also been used as a graphical interface for the modeling of loops in comparative modeling projects, as well as restrained flexible fitting of comparative models into cryo-electron microscopy maps. There are currently many more known protein sequences than there are protein structures, and thus the determination of protein structure from sequence by comparative or homology modeling is of great interest. The MODELLER package is commonly used for this purpose (~11,000 different users have downloaded the package so far) [3]. Two particular areas of current interest are the refinement of protein loops using statistical potentials [4], and the use of additional sources of data, such as from cryo-electron microscopy experiments [5-7]. While MODELLER is a powerful package, it can be hard to use effectively without visual feedback, for instance to see the fit between a density map and a protein model, or to see the configuration of a set of loops. An extension to CHIMERA was created to simplify the use of MODELLER for loop modeling. Given a starting protein structure, the user is able to graphically select a set of residues for further loop refinement. The inputs for MODELLER are then synthesized and a number of candidate loop models built. These can then be viewed and ranked within CHIMERA. In the future, we plan to link MODELLER and Chimera more closely to make the loop modeling process itself more interactive (such that the user can terminate obviously bad loops, 'guide' the optimization by manually perturbing the structure, or adjust the parameters). In addition to loop modeling, this same approach will be applicable to the EM density fitting tools in MODELLER (Mod-EM), and can improve the procedure by which density maps are used to improve the accuracy of comparative models. This exciting project nicely meshes with the visualization tools that the RBVI has been developing for the cryoEM community.References:
- U. Pieper, N. Eswar, H. Braberg, M.S. Madhusudhan, F.P. Davis, A.C. Stuart, N. Mirkovic, A. Rossi, M.A. Marti-Renom, A. Fiser, B. Webb, D. Greenblatt, C.C. Huang, T.E. Ferrin, and A. Sali. "MODBASE, A Database of Annotated Comparative Protein Structure Models, and Associated Resources," Nuc. Acids Res. 32, D217-D222, 2004.
- U. Pieper, N. Eswar, F.P. Davis, H. Braberg, M.S. Madhusudhan, A. Rossi, M. Marti-Renom, R. Karchin, B.M. Webb, D. Eramian, M.Y. Shen, L. Kelly, F. Melo, A. Sali. "MODBASE, A database of annotated comparative protein structure models and associated resources," Nuc. Acids Res. 34, D291-D295, 2006.
- A. Sali, T.L. Blundell. "Comparative protein modelling by satisfaction of spatial restraints," J. Mol. Biol. 234, 779-815, 1993.
- A. Fiser, R.K. Do, A. Sali. "Modeling of loops in protein structures" Protein Sci 9, 1753-1773, 2000.
- M. Topf, M.L. Baker, B. John, W. Chiu, A. Sali. "Structural Characterization of Components of Protein Assemblies by Comparative Modeling and Electron Cryo-Microscop," J. Struct. Biol. 149, 191-203, 2005.
- M. Topf, A. Sali. "Combining Electron Microscopy and Comparative Protein Structure Modeling," Current Opinion in Structural Biology 15, 578-585, 2005.
- M. Topf, M.L. Baker, M.A. Marti-Renom, W. Chiu, A. Sali. "Refinement of Protein Structures by Iterative Comparative Modeling and CryoEM Density Fitting," J. Mol. Biol. 357, 1655-1668, 2006.
Laboratory Overview | Research | Outreach & Training | Available Resources | Visitors Center | Search