

 about
projects
people
publications
resources
resources
visit us
visit us
search
search
about
projects
people
publications
resources
resources
visit us
visit us
search
search
Quick Links
Recent Citations
Molecular mechanism of IgE-mediated FcεRI activation. Chen M, Su Q, Shi Y. Nature. 2025 Jan 9;637(8045):453–460.
High-resolution cryo-EM using a common LaB6 120-keV electron microscope equipped with a sub-200-keV direct electron detector. Venugopal H, Mobbs J et al. Sci Adv. 2025 Jan 3;11(1):eadr0438.
Read-write mechanisms of H2A ubiquitination by Polycomb repressive complex 1. López VG, Valencia-Sánchez MI et al. Nature. 2024 Dec 19;636(8043):755–761.
Gliocidin is a nicotinamide-mimetic prodrug that targets glioblastoma. Chen YJ, Iyer SV et al. Nature. 2024 Dec 12;636(8042):466-473.
Active-reset protein sensors enable continuous in vivo monitoring of inflammation. Zargartalebi H, Mirzaie S et al. Science. 2024 Dec 6;386(6726):1146-1153.
Previously featured citations...Chimera Search
Google™ SearchNews
December 25, 2024

|
October 14, 2024
Planned downtime: The Chimera and ChimeraX websites, web services (Blast Protein, Modeller, ...) and cgl.ucsf.edu e-mail will be unavailable starting Monday, Oct 14 10 AM PDT, continuing throughout the week and potentially the weekend (Oct 14-20).
August 1, 2024
Planned downtime: The Chimera and ChimeraX websites, web services (Blast Protein, Modeller, ...) and cgl.ucsf.edu e-mail will be unavailable August 1, 3-6 pm PDT.
Previous news...Upcoming Events

UCSF Chimera is a program for the interactive visualization and analysis of molecular structures and related data, including density maps, trajectories, and sequence alignments. It is available free of charge for noncommercial use. Commercial users, please see Chimera commercial licensing.
We encourage Chimera users to try ChimeraX for much better performance with large structures, as well as other major advantages and completely new features in addition to nearly all the capabilities of Chimera (details...).
Chimera is no longer under active development. Chimera development was supported by a grant from the National Institutes of Health (P41-GM103311) that ended in 2018.
Feature Highlight

Users can easily import structure-related data into Chimera in the form of attributes, or values associated with atoms, residues, or models. The data can be imported with Define Attribute and then represented visually with color ranges, atomic radii, or "worm" thickness. Such data can also be manipulated programmatically in Chimera, and in fact Chimera was designed with extensibility and programmability in mind. It is largely implemented in Python, with certain features coded in C++ for efficiency. Python is an easy-to-learn interpreted language with object-oriented features. All of Chimera's functionality is accessible through Python and users can implement their own algorithms and extensions without any Chimera code changes, so any such custom extensions will continue to work across Chimera releases. Many programming examples are provided to assist extension writers.
(More features...)Gallery Sample
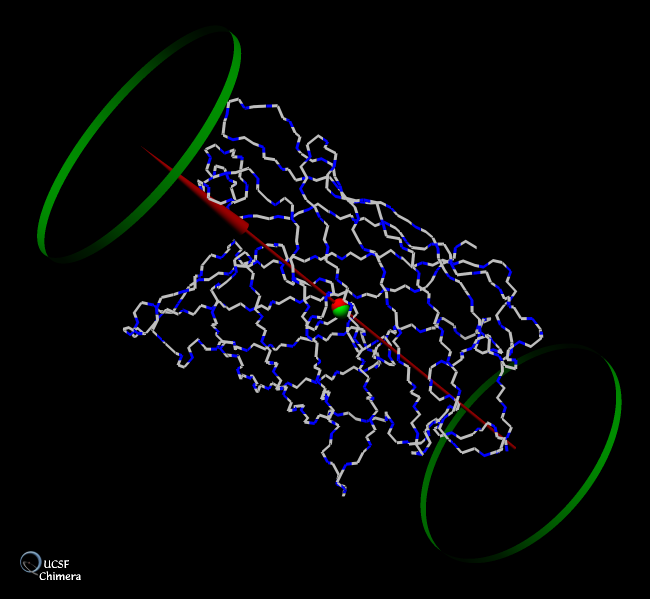
BILD format was used in Chimera to annotate the barrel structure of green fluorescent protein with its centroid, major axis (red arrow), and an enclosing cylinder (shown with green hoops). The BILD file green.bild was generated with the python program green.py using the coordinates in green.pdb. Gallery entry courtesy of Mike Ess, Yeast Resource Center, University of Washington. (More samples...)
About RBVI | Projects | People | Publications | Resources | Visit Us
Copyright 2018 Regents of the University of California. All rights reserved.