


structureViz: Linking Cytoscape and Chimera
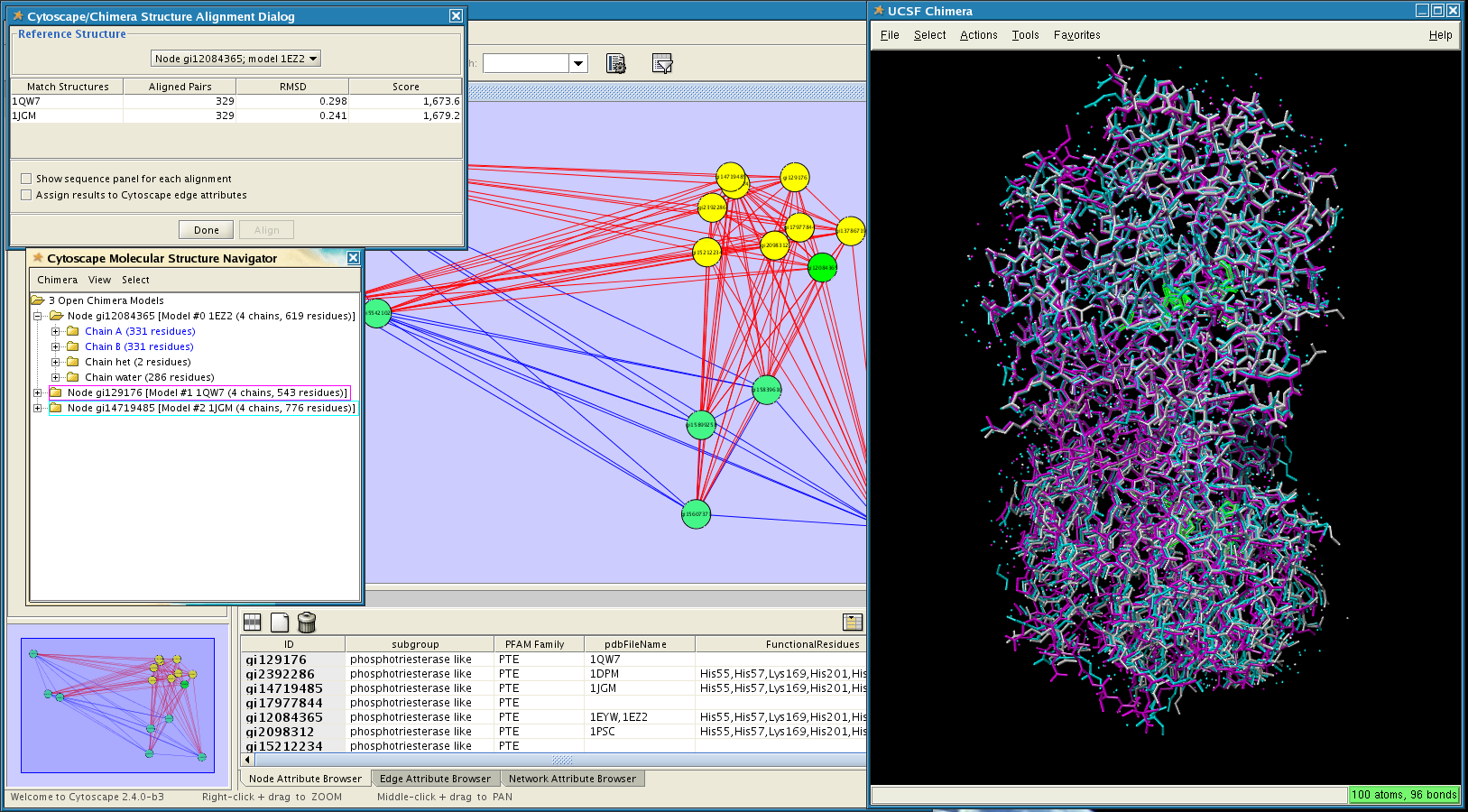
Figure 1. structureViz in action. In this screenshot, the PFAM family PTE (phosphotriesterase) has been opened in Cytoscape. Three of the available structures have been opened in UCSF Chimera and spatially aligned. Parts of one structure (pdb: 1EZ2) are selected, as indicated with green outlines in the Chimera window. Click on the image to enlarge it.
UCSF structureViz is a Cytoscape plugin that links visualization of biological networks in Cytoscape with visualization and analysis of molecular structures in UCSF Chimera. It is part of a broader effort to develop and apply tools for the visualization and analysis of biological context.
Networks have nodes and edges. For structureViz, molecular structures are associated with nodes using the node attributes. Macromolecular structures are specified by Protein Data Bank identifiers (PDB IDs) and small molecule structures by SMILES strings. Homology-modeled protein structures can also be associated, as detailed below.
As an alternative to interacting with Chimera directly, structureViz provides a simplified, tree-based interface to structures called the Cytoscape Molecular Structure Navigator. This interface allows selecting models, chains, and residues and adjusting their display. Basic interfaces are also provided for spatially aligning macromolecular structures and for analyzing steric clashes and hydrogen bonds.
Contents
- Installation
- Adding structure annotations to a Cytoscape network
- structureViz menus
- The Molecular Structure Navigator
- Loading modeled structures
- Aligning structures
- structureViz CyCommands
- Sample data and resources
| [Contents] [Top] |
1. Installation
- If you haven't already done so, download and install UCSF Chimera.
- Install structureViz from the Cytoscape Plugin Manager (see the Manage Plugins option in the Plugins menu; structureViz is listed under the Analysis Plugins category).
- Restart Cytoscape.
- Now structureViz actions should be available in the Cytoscape Plugins menu and node context menus.
| [Contents] [Top] |
2. Adding structure annotations to a Cytoscape network
Annotating a network with structures entails creating new node attributes and populating those attributes with structure identifiers. The type of each attribute can be String (if multiple identifiers, a comma-separated list) or List (with each identifier given as a single string).
- Macromolecular structure (PDB) identifiers. One or more PDB ID codes can be associated with a node as an attribute named Structure, structure, pdb, pdbFileName, PDB ID, or biopax.xref.PDB.
- Small molecule (SMILES) identifiers. One or more SMILES strings can be associated with a node as an attribute named Smiles, smiles, or SMILES. This mainly provides a convenient way to load small molecule structures into Chimera. For more traditional cheminformatics applications, see the chemViz Cytoscape plugin.
- Residue identifiers. An attribute named FunctionalResidues or ResidueList can be used to specify residues of interest. In general, a residue specification is of the form pdbID#residueID.chainID. The pdbID is the 4-character PDB ID of the structure. A chainID is a character used in the structure file to group residues by chain. A residueID may be a single-letter code and residue number such as H263, a three-letter code and number such as His263, or simply a residue number, such as 263. Like the other types of identifiers, residue specifications can be given individually or as a comma-separated list: His236,Lys238, Met240. A range of residues can also be specified: 236-250. To handle cases in which a node is associated with multiple structures and the residues of interest differ between those structures, PDB identifiers can be included: 2uz9#His82,2uz9#Thr102. If no PDB identifier is given, the residue specification is assumed to apply to all of the structures annotated to the node.
In addition, homology-modeled structures can be fetched from ModBase if the node ID is a ModBase-searchable type of sequence identifier and models are available for that sequence (see Loading modeled structures).
| [Contents] [Top] |
3. structureViz menus
structureViz provides two similar sets of menus, one under the Plugins→Sequence/Structure Tools top-level menu and the other as part of the node context menus under Structure Visualization. The structureViz context menus may be accessed by mouse right-click in the Cytoscape network window. The two sets of menus currently present the same options to the user, except that Find modeled structures and Select residues are only available from the node context menu.
- Open structure(s)→
- Allow the user to specify which of the structures associated with the currently selected node(s) should be opened in Chimera. This action is enabled when structureViz detects a node attribute with one of the recognized names for structure annotations (see Adding structure annotations...). PDB entries are fetched over the web from the RCSB PDB. Small molecule structures are generated from SMILES strings using the smi23d web service provided by the Cheminformatics Group at Indiana University. When a structure has been opened in Chimera, the Molecular Structure Navigator will appear.
- Align structures
- Open all structures associated with the currently selected node(s), and if there are at least two, invoke the Cytoscape/Chimera Structure Alignment Dialog (see Aligning structures). (To avoid opening all of the structures, instead open only the structures of interest with the Open structure(s)→ action described above and align them with Chimera→Align Structures→ in the Molecular Structure Navigator top-level menu.)
- Select residues (node context menu only)
- The Select residues menu is present only when the node has a value for the FunctionalResidues or ResidueList attribute. In that case, the relevant structures are opened (if they haven't been already) and the associated residues are selected in both Chimera and the Molecular Structure Navigator.
- Find modeled structures (node context menu only)
- Search ModBase for homology-modeled structures that match the ID for this node. The node ID must be a SwissProt, TrEMBL, GenPept or PIR sequence accession code. If modeled structures are available for the sequence, they are fetched over the web and opened in Chimera (see Loading modeled structures).
- Close structure(s)→
- Allow the user to specify which of the open structures should be removed from both Chimera and the Molecular Structure Navigator.
- Exit Chimera
- Close all structures and quit from UCSF Chimera.
| [Contents] [Top] |
4. The Molecular Structure Navigator
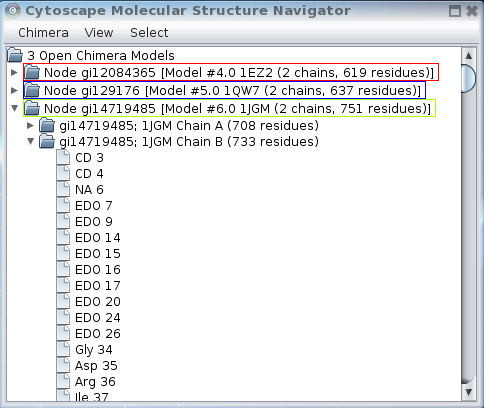
Figure 2. The Molecular Structure Navigator dialog. In this image, the Molecular Structure Navigator shows three open structures (models): 1EZ2, 1QW7, and 1JGM. The entry for 1JGM has been expanded to show the two chains, and the second chain has been expanded to show the initial residues in the PDB entry.
When structureViz is used to open structures in Chimera, the Molecular Structure Navigator appears (see Figure 2). This dialog provides an easy and convenient way to interact with structures regardless of a user's familiarity with Chimera. Each structure has three levels:
- The model level corresponds to a single opened structure. The dialog reports the associated node and the total numbers of chains and residues in the model. The colored outline box reflects the default color of the model in Chimera.
- The chain level corresponds to groups of residues within a model that share a chain identifier.
- The residue level reflects individual residues. Residue names and numbers are shown.
The levels in the hierarchy can be expanded and contracted. Selecting a model, chain, or residue in the Molecular Structure Navigator selects the corresponding structural element in Chimera.
In Figure 2, there are three entries at the model level, reflecting the three open structures. The model entry for 1JGM has been expanded to show the two chains that are part of this model (phosphotriesterase is a dimer), and the second chain (B) has been expanded to show the residues contained within that chain.
In addition to selection and navigation within the structural hierarchy, the Molecular Structure Navigator provides menus (top-level and context) for aligning and analyzing structures and adjusting their display. It serves as an alternative interface to UCSF Chimera, but the standard Chimera menus, tools, and commands can also be employed.
| [Contents] [Top] |
4.1 Top-level menus
The menu bar across the top of the Molecular Structure Navigator contains the following:
- Chimera→
- Align structures→
- Invoke the Cytoscape/Chimera Structure Alignment Dialog for aligning (superimposing) structures by model or by chain. In either case, the user must indicate one reference model (or chain) and one or more other models (or chains) to match to the reference. Results from each pairwise match can be used to create new edges in the original Cytoscape network (see Aligning structures).
- Focus all
- Zoom out to include all structures in the UCSF Chimera display.
- Presets→
-
Presets are predefined combinations of display settings. Choosing a preset
from the menu applies its settings.
The interactive presets are meant for
interactive manipulation and analysis.
They may change what items (atoms, ribbons, surfaces) are displayed and how
they are colored, and set the background color to black.
The publication presets are for generating
images for presentation and publication.
They do not change what items are displayed or their colors,
but may change their styles, and set the background color to white.
Publication presets may decrease interactive performance because finer
divisions are used to render atoms, ribbons, and surfaces.
See also:
tips on preparing images in Chimera
- interactive 1 (ribbons)
- interactive 2 (all atoms)
- interactive 3 (hydrophobicity surface)
- publication 1 (silhouette, rounded ribbon, sticks)
- publication 2 (silhouette, licorice, sticks)
- publication 3 (depth-cued, rounded ribbon, sticks)
- publication 4 (depth-cued, licorice, sticks)
- Clash detection→
-
Identify steric conflicts (close contacts) involving the selected atoms
and show them with lines.
- Find all clashes
- Identify clashes between the selected atoms and all other atoms.
- Find clashes within models
- Identify clashes between the selected atoms and other atoms in the same model.
- Clear clashes
- Remove lines showing clashes.
- Hydrogen bond detection→
-
Find hydrogen bonds→
identifies hydrogen bonds involving the currently selected atoms,
restricted as follows:
- Between models - inter-model hydrogen bonds only (note that different chains in the same structure are still in the same model)
- Within models - intra-model hydrogen bonds only
- Both - unrestricted; both inter- and intra-model hydrogen bonds
- Exit
- Quit from UCSF Chimera and close the Molecular Structure Navigator.
- View→
- Collapse model tree
- Collapse models to hide residues and chains in the Molecular Structure Navigator tree.
- Expand all models
- Expand models to show all chains in the Molecular Structure Navigator tree.
- Expand all chains
- Expand to show all chains and residues in the Molecular Structure Navigator tree.
- Refresh model tree
- Refresh model tree
- Refresh model tree
- Refresh the Molecular Structure Navigator tree.
- Show residues as...→
-
Set how amino acids are listed in the Molecular Structure Navigator:
- single letter
- Use single-letter abbreviations for amino acid residues.
- three letters
- Use three-letter abbreviations for amino acid residues.
- full name
- Use the full names of amino acid residues.
- Select→
- Select parts of structures in UCSF Chimera and the Molecular Structure Navigator.
- Protein
- Select all amino acid residues.
- Nucleic acid
- Select all nucleic acid residues.
- Ligand
- Select all ligand residues.
- Ions
- Select all monatomic ions.
- Solvent
- Select all solvent residues.
- Secondary structure→
-
Select amino acid residues according to their secondary structure assignments:
- Helix
- Strand
- Coil (non-helix, non-strand amino acid residues)
- Functional Residues
- Select functional residues (available when the appropriate node attribute has been assigned).
- Invert selection
- Invert the current selection (select the unselected atoms and deselect the selected atoms).
- Clear selection
- Clear the selection (deselect all atoms).
| [Contents] [Top] |
4.2 Context menus
Two categories of context menus can be accessed by mouse right-click in the Molecular Structure Navigator: those with generic actions, and those which also include special actions depending on the context level (model, residue, or chain).
| [Contents] [Top] |
- 4.2.1 Generic actions
-
- Show→ Control the display of atoms and bonds in the current context.
-
- All
- Show all atoms.
- Backbone only
- Show backbone atoms only (these may be obscured by ribbons).
- Sequence
- Show the sequence of each chain in a separate window (Chimera's Sequence tool). In the residue context, the entire chain sequence will still be shown, but with any residue(s) selected in Chimera and the Molecular Structure Navigator highlighted in bright green.
- Hide
- Hide all atoms.
- Focus
- Adjust the view and center of rotation in Chimera to focus on the models, chains, or residues specified by the context.
- Color→
-
- Rainbow by chain
- (only available in the model context) Assign each chain in the model a separate color.
- Rainbow by residue
- (only available in the model or chain context) Rainbow-color-code the residues in each chain, starting from blue and ending at red.
- Atoms/Bonds→
-
Change atom/bond color.
- By element
- Color-code atoms by element: grey for carbon, red for oxygen, blue for nitrogen, yellow for sulfur, etc. (see element colors).
- By heteroatom
- Use the model default color for carbon, color-code other atoms by element.
- Ribbons→
- Change ribbon color.
- Surfaces→
- Change surface color.
- Labels→
- Change label color.
- Depict→ Control display styles. Different styles can serve to highlight different aspects of a structure, from overall shape to specific atomic interactions.
-
- Wire
- Show bonds as wires and atoms as dots; this style is the simplest and least computationally demanding.
- Stick
- Show bonds as sticks and atoms as endcaps (rounded ends).
- Ball & Stick
- Show bonds as sticks and atoms as balls.
- Sphere or CPK
- Show atoms as spheres using their VDW radii.
- Ribbon→
- A ribbon is a cartoon-like representation that traces the
backbone of a protein or nucleic acid. It is particularly useful for
viewing the secondary structure of a protein.
- Hide
- Hide ribbon.
- Flat
- Show flat ribbon.
- Edged
- Show edged ribbon (rectangular cross-section).
- Round
- Show rounded ribbon (rounded cross-section).
- Surface→
- Molecular surfaces highlight the overall shapes of structures and
their binding pockets.
- Hide
- Hide molecular surface.
- Solid
- Show surface as solid. To reveal underlying objects, adjust Transparency (below).
- Mesh
- Show surface as mesh.
- Dot
- Show surface as dots.
- Transparency→
- Adjust surface transparency.
- Label→
-
- Hide
- Hide atom and residue labels.
- Atom name
- Label atoms by name.
- Element
- Label atoms by element symbol.
- IDATM type
- Label atoms by IDATM type.
- Residue
- Label residues by identifier.
- Clear selection
- Clear the selection (deselect all atoms). This action applies to all selected items, regardless of the context of the pop-up.
- Delete selection
- Delete all atoms in the current selection. This action applies to all selected items, regardless of the context of the pop-up. Note that deleted atoms cannot be restored except by reopening the original structure, and that deletion will also remove any corresponding ribbon or surface.
Generic actions are always available, although the scope of the action depends on the context of the pop-up menu. When the mouse cursor is placed on a specific item (model, chain, or residue) when the menu is popped up, the action applies specifically to that item. If the mouse pointer is placed over an empty area, the action applies to any selected models, chains, or residues. Note that when nothing is selected, the context menu can not be popped up in an empty area.
| [Contents] [Top] |
- Close model(s)
- Close all of the models in the current context. This will remove them from the Molecular Structure Navigator as well as closing them in Chimera
- Select→
-
Select parts of the models in the current context.
- Protein
- Select all amino acid residues.
- Nucleic acid
- Select all nucleic acid residues.
- Ligand
- Select all ligand residues.
- Ions
- Select all monatomic ions.
- Solvent
- Select all solvent residues.
- Secondary structure→
-
Select amino acid residues according to their secondary structure assignments:
- Helix
- Strand
- Coil (non-helix, non-strand amino acid residues)
- Functional Residues
- Select functional residues (available when the appropriate node attribute has been assigned).
| [Contents] [Top] |
5. Loading modeled structures
If the ID of a node is a SwissProt, TrEMBL, GenPept or PIR sequence accession code, ModBase can be searched for homology-modeled structures of the corresponding sequence using Find modeled structures in the node context menu. If ModBase contains modeled structures of that sequence, they are fetched over the web and opened in Chimera. Chimera also displays a dialog, the ModBase Model List, that lists the models and some associated information. There may be a large number of modeled structures, whereas a user may want only one. Clicking a row in the ModBase Model List shows just the corresponding model (hides the others in the list), and the scores and other information in the list can help to identify an appropriate model for further analysis. To close all other models in Chimera, select the desired model in the Molecular Structure Navigator, invert the selection (Select→Invert selection), and then use Close model(s) in the model context menu.
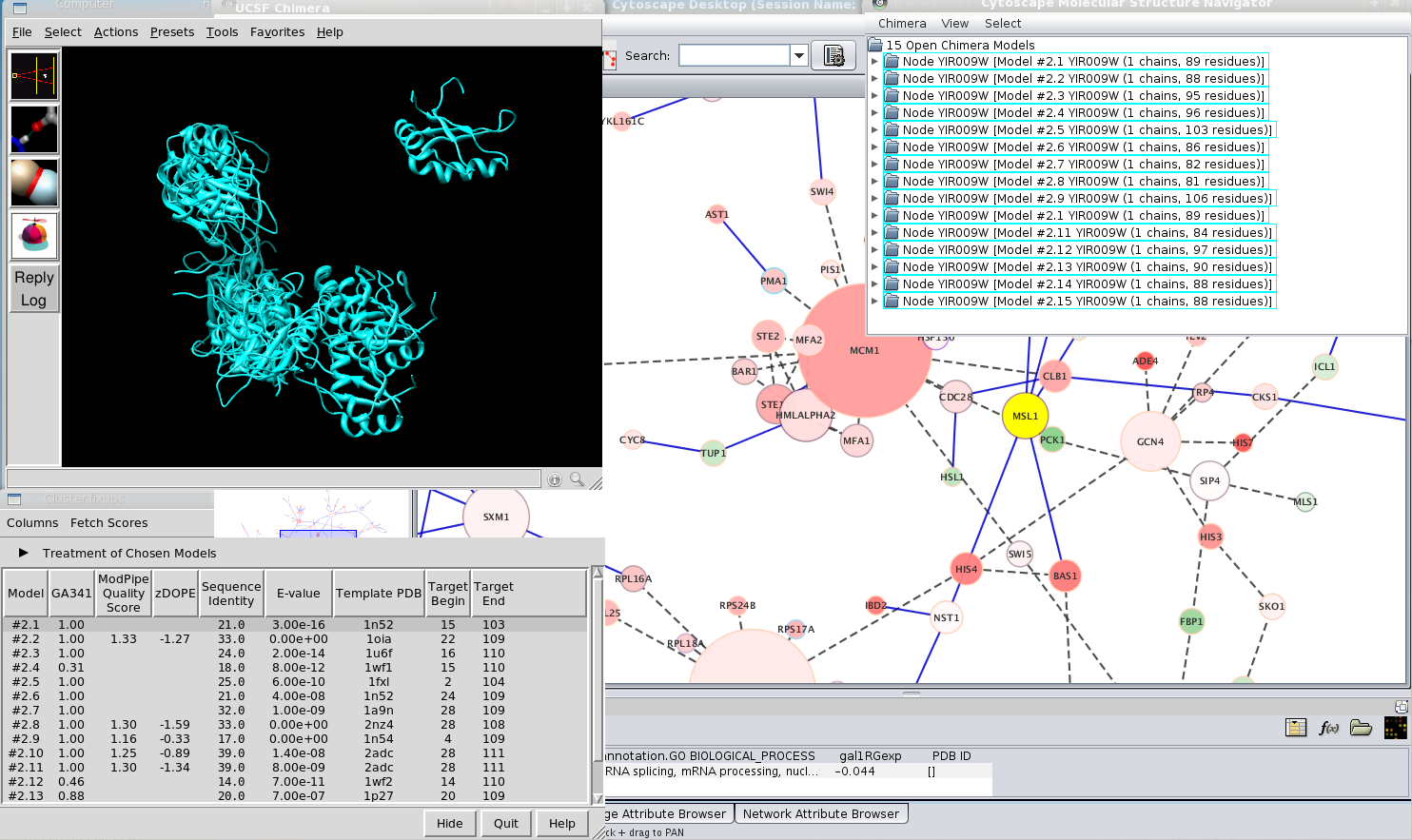
Figure 3. Importing modeled structures. In this image, the Molecular Structure Navigator shows the result of finding modeled structures for node MSL1, which has the ID YIR009W. UCSF Chimera has queried ModBase and retrieved the results.
| [Contents] [Top] |
6. Aligning structures
Structures can be spatially aligned (superimposed) and the corresponding alignment scores assigned to edges in the Cytoscape network. The Cytoscape/Chimera Structure Alignment Dialog can be invoked from the structureViz menu or the Molecular Structure Navigator top-level menu. In the latter menu, Align structures includes suboptions by chain for choosing specific chains to use in the alignment, versus by model for choosing whole models.
In the dialog, it is necessary to specify one reference model (chain, if aligning by chain) and one or more models (chains) to match. Each structure to match is superimposed on the reference structure by first creating a sequence alignment and then fitting the aligned residue pairs. The sequence alignment is constructed using a score based both on residue type (as in a traditional sequence alignment) and on secondary structure assignments (preferentially aligning helix with helix and strand with strand). If models were chosen rather than specific chains, all pairwise combinations of chains in the two models are aligned and the chain pair with the best score is used for the fit. The fit is iterated to remove far-apart residue pairs so that only a well-superimposed structural core is used for the final fit. (For details, see the Chimera MatchMaker documentation; default alignment and matching settings are used.) Additional dialog options:
- Show sequence panel for each alignment - whether Chimera should display the sequence alignment used to superimpose each pair of structures. In the sequence alignment window (Chimera's Multalign Viewer), the residue pairs used in the final fit are indicated with light orange boxes.
- Assign results to Cytoscape edge attributes - whether to add an edge between the nodes associated with each reference-match pair of structures, and to assign the alignment scores as attributes of that edge. Alignment Pairs is the number of residue pairs used in the final fit. RMSD is the α-carbon RMSD of that set of residues in the final fit. Score is the score of the sequence alignment (based on both residue types and secondary structure as described above) used to align the structures.
Users should be aware that the scores may not correlate well with structural similarity or superposition quality when the sequences are dissimilar and hard to align. Successful superposition only requires the sequence alignments to be partly correct, as incorrect regions tend to be omitted during fit iteration. Omitting parts of the sequence alignment from the fit will affect the number of residue pairs used and the RMSD, even if the resulting superposition is entirely correct. However, in typical usage, the proteins are similar and their sequences easy to align.
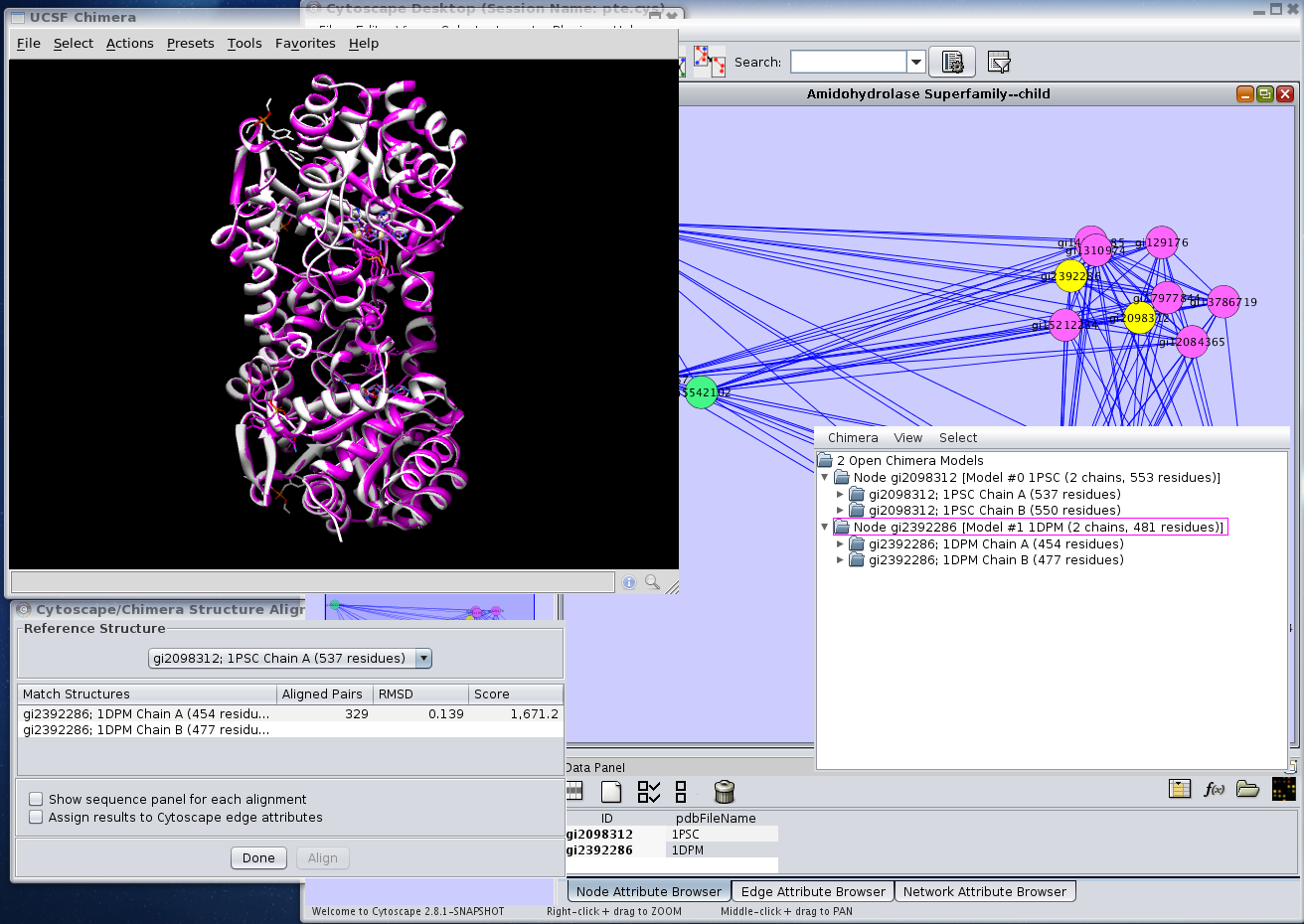
Figure 4. Using UCSF Chimera's MatchMaker capabilities to perform an alignment of two structures. In this image two structures: 1PSC and 1DPM had been loaded into Chimera and aligned (Chimera→Align structures→by chain).
| [Contents] [Top] |
7. structureViz CyCommands
In order to facilitate the use of structureViz and UCSF Chimera from other plugins, the structureViz plugin exports a number of commands using the CyCommand mechanism. These commands may be used by other plugins, or as part of scripts that are executed using the commandTool plugin.
There are a number of arguments which are repeated for several of the commands. In particular: structurelist, atomspec, and structuretype are defined here and referenced from each of the commands outlined below:
- structurelist
- This a list of structures which may be specified as either a list of open PDB identifiers, model numbers,
or Cytoscape nodes that are associated with open structures. As with other lists, these values are comma-separated
and enclosed in quotes. Examples:
- "1tup,1gzh,1tsr"
- "#0,#2"
- "tp53"
- selected
- all
- atomspec
- An atomspec is the specification for an atom in Chimera, however structureViz will
only allow specification to the residue level. The general form of the atomspec is:
- #model.submodel:residuelist.chain
- structuretype
- This argument is used to control the selection or display of structural components based on
the class or chemical attribute of the component. The legal keywords are:
- full: Display or select the full backbone.
- minimal: Display or select the minimal backbone.
- ions: Display or select ions
- ligand: Display or select ligands
- main: Display or select the main structure
- nucleic acid: Display or select nucleic acids
- protein: Display or select proteins
- solvent: Display or select the solvent
- helix: Display or select components with helical secondary structure
- strand: Display or select components with strand (or sheet) secondary structure
- coil: Display or select components that are neither helix nor strand
- with CA/C1': Display or select atoms in side chains including the CA (for amino acids) and C1' (for nucleic acids)
- without CA/C1': Display or select atoms in side chains excluding the CA and/or C1'
In general, all of the commands include several arguments, which are described below the command description. The arguments and their values are specified as name, value pairs separated by an equals sign (=). For example, to align two structures, the user might enter:
align structures reference="1tup" structurelist="1gzh,1tsr" assignattributes=false showsequences=trueNote that the text arguments are placed within quotes.
If the argument has a default value, that value is shown below in brackets ([]). Literal values, such as the booleans true and false or the keyword selected are shown in bold, while descriptions of the argument type such as atomSpec or chainSpec are shown in italics, usually with an example following in parentheses.
- structureviz align chains
-
Perform sequence-driven structural superposition on a group of structures by chain.
Arguments:
- referencechain=chainSpec: This is the chain to be used as the reference for the pairwise alignments. The format is the same as an atomspec except that only the chain component is used.
- chainlist=chainlist : The list of chains to align to the referencechain, where each element in the list is an atomspec from which only the model and chain component are extracted.
- showsequences=[false]: If true, the sequence alignment for each pair of structures is shown.
- createedges=[false]: If true new edges will be created in the Cytoscape network that represent the similarity of each structure with the reference chain.
- assignattributes=[true]: If true the RMSD, Alignment Score, and number of aligned pairs are added to the Cytoscape network as attributes.
- structureviz align structures
-
Perform sequence-driven structural superposition on a group of structures.
Arguments:
- reference=structure: This is the structure to be used as the reference for the pairwise alignments. The format is the same as an structurelist except that only a single structure may be specified.
- structurelist=structurelist : The list of structures to align to the reference structure.
- showsequences=[false]: If true, the sequence alignment for each pair of structures is shown.
- createedges=[false]: If true new edges will be created in the Cytoscape network that represent the similarity of each structure with the reference chain.
- assignattributes=[true]: If true the RMSD, Alignment Score, and number of aligned pairs are added to the Cytoscape network as attributes.
- structureviz clear clashes
-
Clear the clashes shown in the display and disable clash detection.
No arguments
- structureviz clear hbonds
-
Clear the hydrogen bonds shown in the display.
No arguments
- structureviz clear selection
-
Clear all selections.
No arguments
- structureviz close
-
Close some or all of the currently opened structures.
Arguments:
- structurelist=[selected]: This is the list of structures (in the form of a structurelist to be closed.
- structureviz color
-
Color part or all of a structure. This command allows the user to specify the
color for residues, labels, ribbons, and surfaces. The color
is specified as a recognized Chimera
color name or
a hexidecimal RGB value: #rrggbb.
Arguments:
- preset=preset_type: One of the recognized Chimera presets (e.g. preset="interactive 1"). If this argument is used, no structurelist or atomspec argument is required (and if provided, is ignored).. All other color arguments require either a sturcturelist or an atomspec, or an atomspec of "selected" is assumed.
- residues=color: set the color of the targeted residues to color.
- labels=color: set the color of the targeted labels to color.
- ribbons=color: set the color of the targeted ribbons to color.
- surfaces=color: set the color of the targeted surfaces to color.
- structurelist=structurelist: This is the list of structures (in the form of a structurelist to apply the specified colors to.
- atomspec=atomspec: This is the list of models, chains, or residues (in the form of a list of atomspecs to apply the specified colors to.
- structureviz depict
-
Change the depiction (display style) of a structure.
Arguments:
- preset=preset_type: One of the recognized Chimera presets (e.g. preset="interactive 1"). If this argument is used, no structurelist or atomspec argument is required (and if provided, is ignored).. All other depict arguments require either a sturcturelist or an atomspec, or an atomspec of "selected" is assumed.
- style=[none|wire|stick|bs|sphere|cpk]: This is the style of depiction to use for residues in the specified structurelist or atomspec argument, but does not change the visibility of those items. If all of the resides of a structure are hidden (for example because a ribbon representation is being used) this command will appear to have no effect. You will need to first show the sidechains for those residues.
- ribbonstyle=[none|flat|edged|round]: This is the style of depiction to use for ribbons in the specified structurelist or atomspec argument. Unlike the style argument setting the ribbonstyle acts to display the ribbon for the selected structural elements. Setting the ribbonstyle to none hides the ribbon.
- surfacestyle=[none|solid|mesh|dot]: This is the style of depiction to use for surfaces in the specified structurelist or atomspec argument. Like ribbonstyle this argument causes the surface to be calculated and displayed.
- structurelist=structurelist: This is the list of structures (in the form of a structurelist to change the depiction of.
- atomspec=atomspec: This is the list of models, chains, or residues (in the form of a list of atomspecs to change the depiction of.
- structureviz exit
-
Exit Chimera.
No arguments
- structureviz find clashes
-
Find clashes between two models or parts of models.
Arguments:
- structurelist=structurelist: This is the list of structures (in the form of a structurelist to detect classed between.
- atomspec=[selected]: This is the list of models, chains, or residues (in the form of a list of atomspecs to detect clashed between.
- continuous=[true]: This argument tells Chimera to continually update the clash detection and the display of the clashing residues.
- structureviz find hbonds
-
Find hydrogen bonds between two models or parts of models.
Arguments:
- structurelist=structurelist: This is the list of structures (in the form of a structurelist to look for hydrogen bonds between.
- atomspec=[selected]: This is the list of models, chains, or residues (in the form of a list of atomspecs to look for hydrogen bonds betwee.
- structureviz focus
-
Focus the Chimera display on a structure or part of a structure. If no arguments are supplied, the entire
scene is focused.
Arguments:
- structurelist=structurelist: This is the list of structures (in the form of a structurelist to focus on.
- atomspec=atomspec: This is the list of models, chains, or residues (in the form of a list of atomspecs to focus on.
- structureviz hide
-
Hide parts of a structure.
Arguments:
- structurelist=structurelist: This is the list of structures (in the form of a structurelist to hide.
- atomspec=[selected]: This is the list of models, chains, or residues (in the form of a list of atomspecs to hide.
- structuretype=structuretype: The structure type to narrow the selection to or to implicitly select for hiding.
- structureviz list chains
-
List the chains in a structure.
Arguments:
- structurelist=[all]: This is the list of structures (in the form of a structurelist to list the chains for.
- structureviz list residues
-
List the residues in a structure.
Arguments:
- structurelist=[all]: This is the list of structures (in the form of a structurelist to list the residues for.
- chainlist=chainlist: The list of chains to show residues for, where each element in the list is an atomspec from which only the model and chain component are extracted.
- structureviz list selected chains
-
List all of the currently selected chains in Chimera.
Arguments:
- structurelist=[all]: This is the list of structures (in the form of a structurelist to list the selected chains for.
- structureviz selected models
-
List all of the currently selected models in Chimera.
No arguments
- structureviz list selected residues
-
List the currently selected residues in Chimera.
Arguments:
- structurelist=[all]: This is the list of structures (in the form of a structurelist to list the selected residues for.
- chainlist=chainlist: The list of chains to show selected residues for, where each element in the list is an atomspec from which only the model and chain component are extracted.
- structureviz list structures
-
List all of the open structures.
No arguments
- structureviz move
-
Move (translate) a model.
Arguments:
- x=x location: The x coordinate to move the model to.
- y=y location: The y coordinate to move the model to.
- z=z location: The z coordinate to move the model to.
- structurelist=[selected]: This is the list of structures (in the form of a structurelist to be moved.
- structureviz open structure
-
Open a new structure in Chimera.
Arguments:
- pdbid=pdbid: The ID of the structure in the PDB. This will cause Chimera to fetch the designated structure from the PDB.
- modbaseid=modbaseid: The ID of a modeled structure in ModBase. Chimera will fetch the modeled structure from ModBase and display it.
- nodelist=nodelist: A list of nodes in Cytoscape to attempt to find attributes with structural identifiers and open all the identifiers for those nodes.
- showdialog=[false]: if true show the Molecular Structure Navigator dialog after opening the structure in Chimera.
- structureviz rainbow
-
Color part or all of a structure in a rainbow scheme.
Arguments:
- structurelist=structurelist: This is the list of structures (in the form of a structurelist to color in a rainbow.
- atomspec=[selected]: This is the list of models, chains, or residues (in the form of a list of atomspecs to color in a rainbow.
- structureviz rotate
-
Rotate a model.
Arguments:
- x=angle: The amount (in angles) to turn around the x axis.
- y=angle: The amount (in angles) to turn around the y axis.
- z=angle: The amount (in angles) to turn around the z axis.
- center=centerspec: The center of the rotation. This may be one of the keywords: view, models, or front or an x,y,z value representing a point in the scene, or an atomspec for designating a portion of the structure to rotate around.
- structurelist=[selected]: This is the list of structures (in the form of a structurelist to be rotated.
- structureviz select
-
Select a structure or parts of a structure.
Arguments:
- structurelist=structurelist: This is the list of structures (in the form of a structurelist to select.
- atomspec=[selected]: This is the list of models, chains, or residues (in the form of a list of atomspecs to select.
- structuretype=structuretype: The structure type to select.
- structureviz send
-
Send a command to Chimera.
Arguments:
- command=chimera command: The command to send to Chimera. No special parsing or handling is done on the string. For a list of Chimera commands and syntax, see the Chimera User's Guide.
- structureviz show
-
Show parts of a structure.
Arguments:
- structurelist=structurelist: This is the list of structures (in the form of a structurelist to show.
- atomspec=[selected]: This is the list of models, chains, or residues (in the form of a list of atomspecs to show.
- structuretype=structuretype: The structure type to narrow the selection to or to implicitly select for displaying.
| [Contents] [Top] |
8. Sample data and resources
- structureViz tutorial
- small sample network annotated with PDB structures: pte.xgmml (phosphotriesterases)
- larger sample network annotated with PDB structures: AHCircle.xgmml (the amidohydrolase superfamily from the Structure-Function Linkage Database)
- a node attribute file providing known linkages between GI numbers and protein structures: structures.noa
- structureViz publication:
structureViz: Linking Cytoscape and UCSF Chimera. Morris JH, Huang CC, Babbitt PC, Ferrin TE. Bioinformatics 2007. [PMID:17623706]